

A unified catalog of 204,938 reference genomes from the human gut microbiome
- PMID: 32690973
- PMCID: PMC7801254
- DOI: 10.1038/s41587-020-0603-3
A unified catalog of 204,938 reference genomes from the human gut microbiome
Abstract
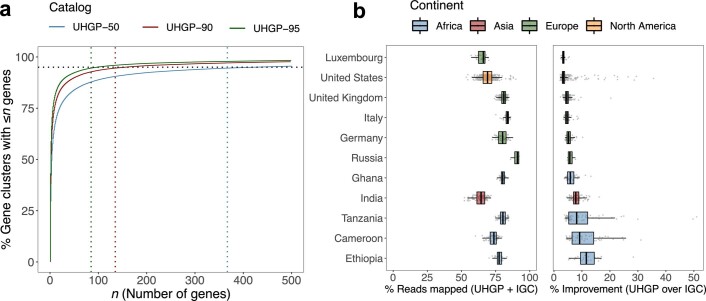
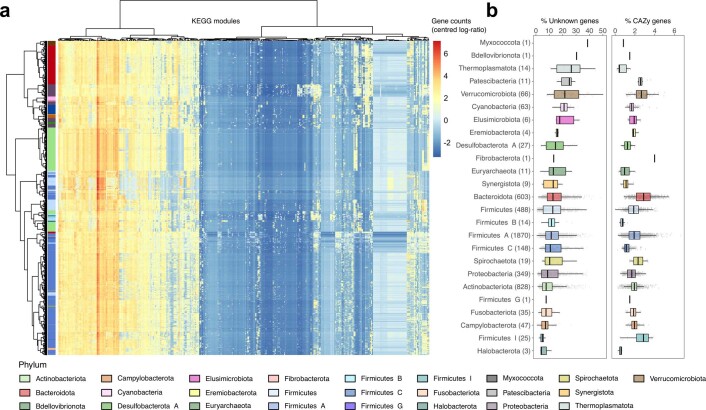
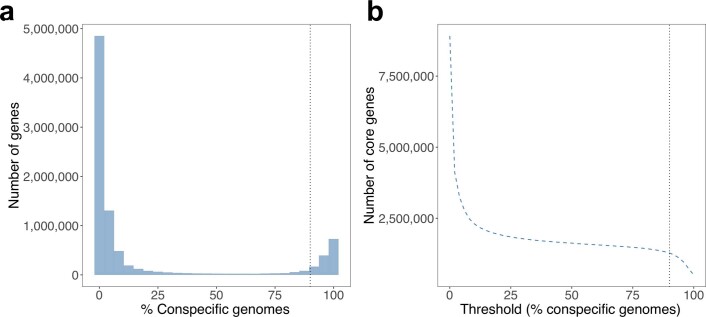
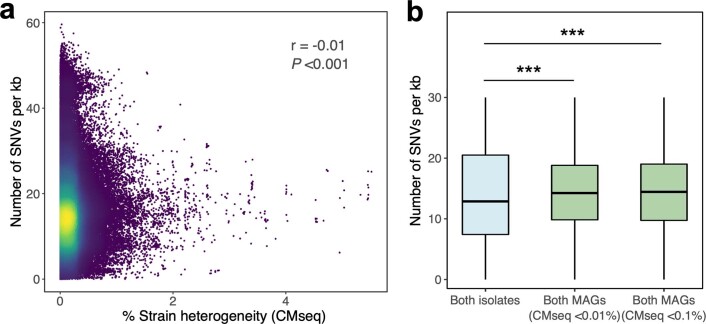
Comprehensive, high-quality reference genomes are required for functional characterization and taxonomic assignment of the human gut microbiota. We present the Unified Human Gastrointestinal Genome (UHGG) collection, comprising 204,938 nonredundant genomes from 4,644 gut prokaryotes. These genomes encode >170 million protein sequences, which we collated in the Unified Human Gastrointestinal Protein (UHGP) catalog. The UHGP more than doubles the number of gut proteins in comparison to those present in the Integrated Gene Catalog. More than 70% of the UHGG species lack cultured representatives, and 40% of the UHGP lack functional annotations. Intraspecies genomic variation analyses revealed a large reservoir of accessory genes and single-nucleotide variants, many of which are specific to individual human populations. The UHGG and UHGP collections will enable studies linking genotypes to phenotypes in the human gut microbiome.
Conflict of interest statement
F.S. is an employee of Enterome. P.H. is a cofounder and is director of Microba Life Sciences Ltd. D.H.P. is a consultant to Microba Life Sciences Ltd. R.D.F. is a consultant to Microbiotica Pty Ltd.
Figures
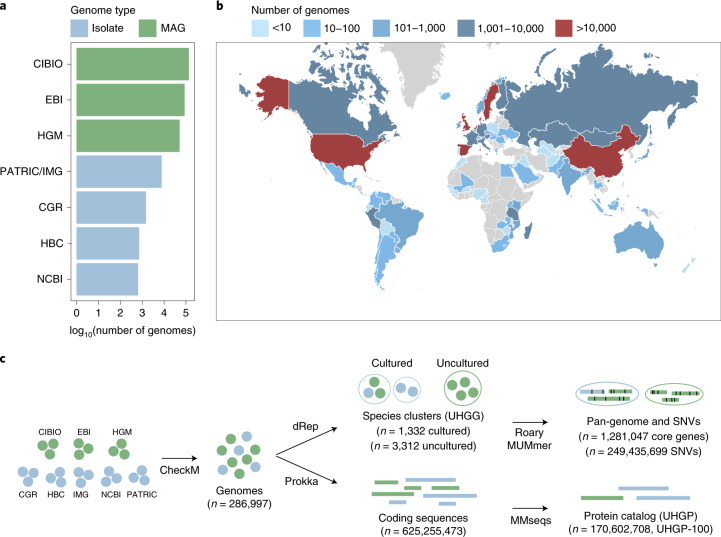
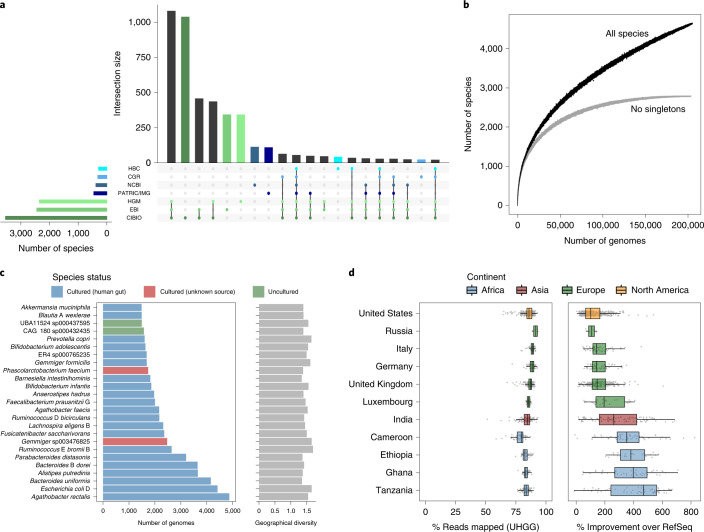
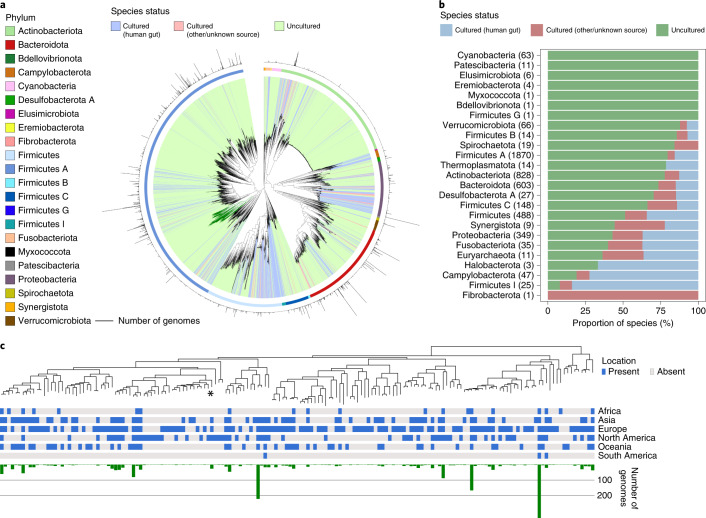
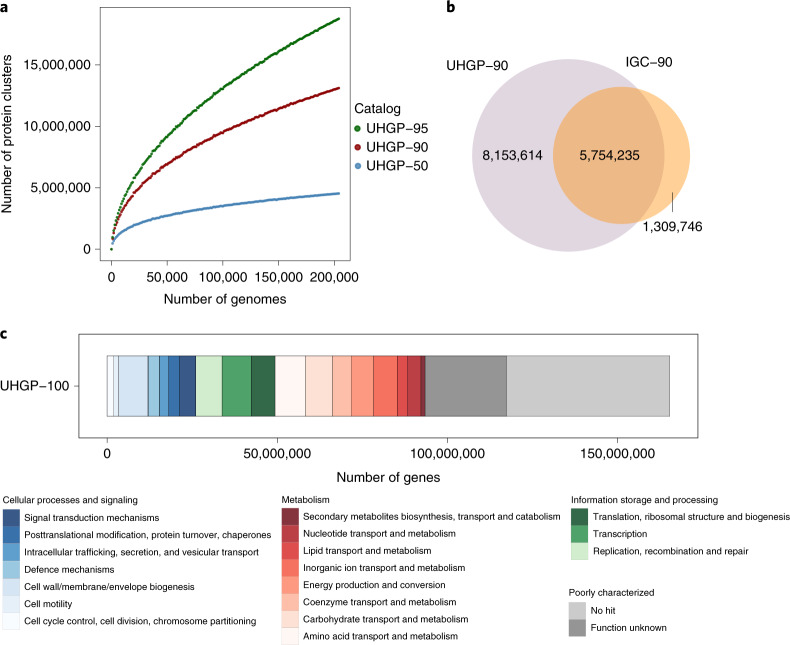
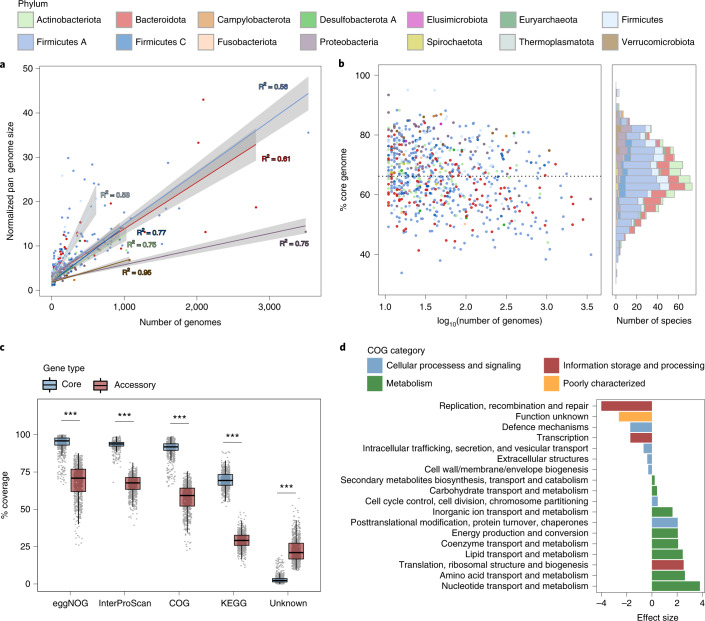
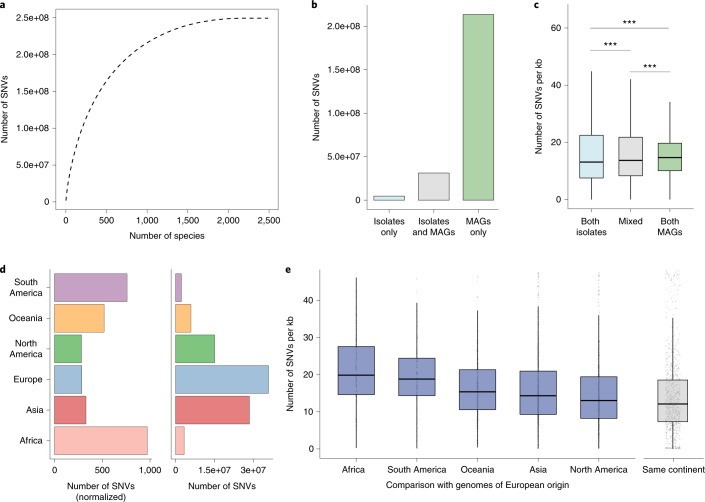
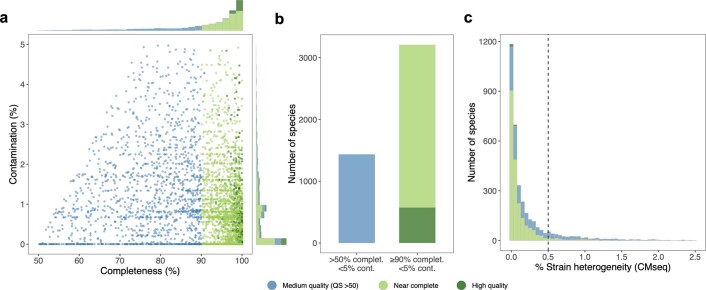
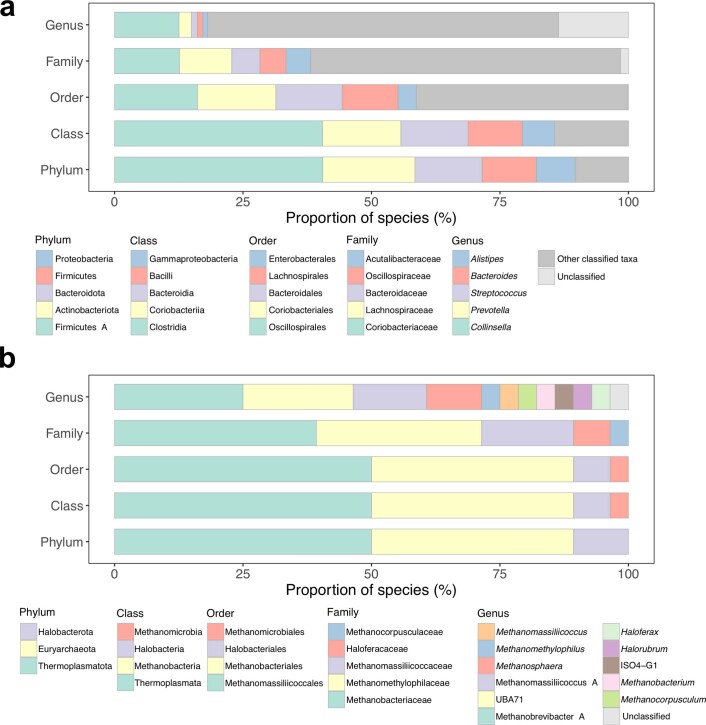
References
-
- Qin J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. - PubMed
-
- Feng Q, et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015;6:6528. - PubMed
-
- Li J, et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32:834–841. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
