

Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2
- PMID: 32694143
- PMCID: PMC7374062
- DOI: 10.1128/mBio.01610-20
Discovery and Genomic Characterization of a 382-Nucleotide Deletion in ORF7b and ORF8 during the Early Evolution of SARS-CoV-2
Abstract
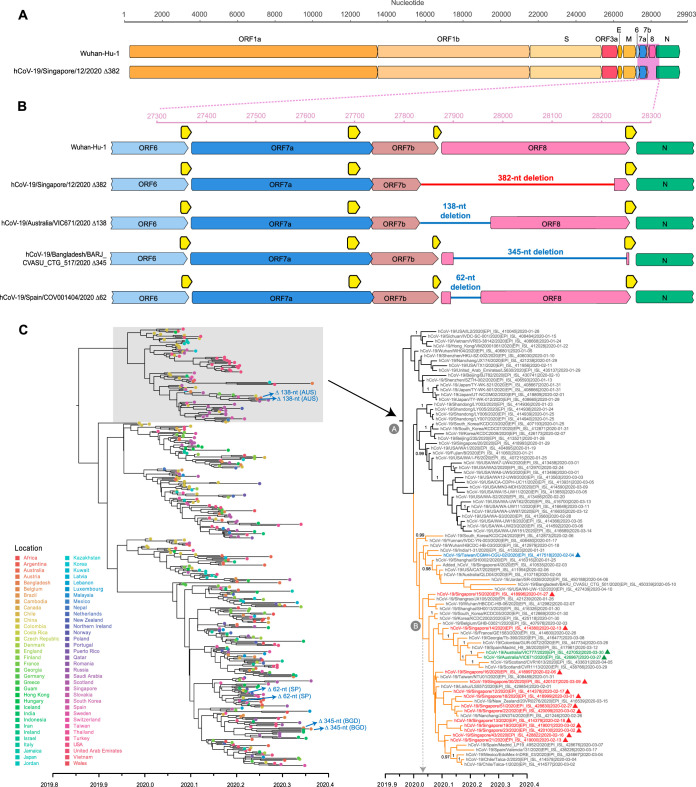
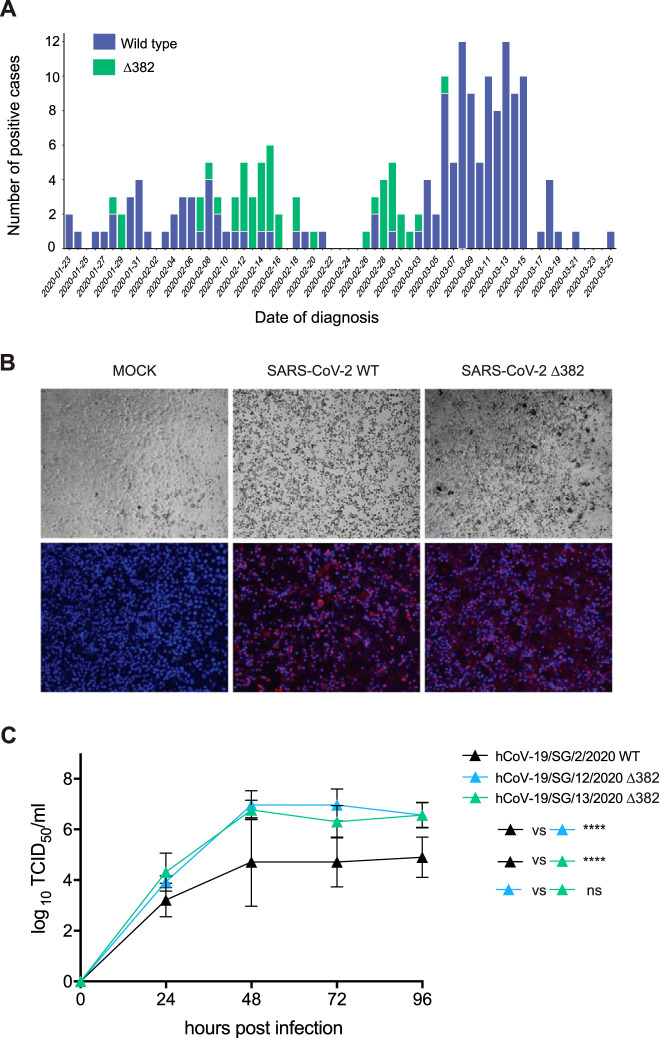
To date, limited genetic changes in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome have been described. Here, we report a 382-nucleotide (nt) deletion in SARS-CoV-2 that truncates open reading frame 7b (ORF7b) and ORF8, removing the ORF8 transcription regulatory sequence (TRS) and eliminating ORF8 transcription. The earliest 382-nt deletion variant was detected in Singapore on 29 January 2020, with the deletion viruses circulating in the country and accounting for 23.6% (45/191) of SARS-CoV-2 samples screened in this study. SARS-CoV-2 with the same deletion has since been detected in Taiwan, and other ORF7b/8 deletions of various lengths, ranging from 62 nt to 345 nt, have been observed in other geographic locations, including Australia, Bangladesh, and Spain. Mutations or deletions in ORF8 of SARS-CoV have been associated with reduced replicative fitness and virus attenuation. In contrast, the SARS-CoV-2 382-nt deletion viruses showed significantly higher replicative fitness in vitro than the wild type, while no difference was observed in patient viral load, indicating that the deletion variant viruses retained their replicative fitness. A robust antibody response to ORF8 has been observed in SARS-CoV-2 infection, suggesting that the emergence of ORF8 deletions may be due to immune-driven selection and that further deletion variants may emerge during the sustained transmission of SARS-CoV-2 in humans.IMPORTANCE During the SARS epidemic in 2003/2004, a number of deletions were observed in ORF8 of SARS-CoV, and eventually deletion variants became predominant, leading to the hypothesis that ORF8 was an evolutionary hot spot for adaptation of SARS-CoV to humans. However, due to the successful control of the SARS epidemic, the importance of these deletions for the epidemiological fitness of SARS-CoV in humans could not be established. The emergence of multiple SARS-CoV-2 strains with ORF8 deletions, combined with evidence of a robust immune response to ORF8, suggests that the lack of ORF8 may assist with host immune evasion. In addition to providing a key insight into the evolutionary behavior of SARS-CoV-2 as the virus adapts to its new human hosts, the emergence of ORF8 deletion variants may also impact vaccination strategies.
Keywords: COVID-19; ORF8; natural selection; phylogeny; vaccines.
Copyright © 2020 Su et al.
Figures
References
-
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. doi:10.1016/S0140-6736(20)30251-8. - DOI - PMC - PubMed
-
- World Health Organization. 2020. Coronavirus disease (COVID-2019) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situatio.... Accessed 2 March 2020.
-
- Pung R, Chiew CJ, Young BE, Chin S, Chen MI-C, Clapham HE, Cook AR, Maurer-Stroh S, Toh MPHS, Poh C, Low M, Lum J, Koh VTJ, Mak TM, Cui L, Lin RVTP, Heng D, Leo Y-S, Lye DC, Lee VJM, Singapore 2019 Novel Coronavirus Outbreak Research Team. 2020. Investigation of three clusters of COVID-19 in Singapore: implications for surveillance and response measures. Lancet 395:1039–1046. doi:10.1016/S0140-6736(20)30528-6. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
