

Genetic architecture of complex traits and disease risk predictors
- PMID: 32694572
- PMCID: PMC7374622
- DOI: 10.1038/s41598-020-68881-8
Genetic architecture of complex traits and disease risk predictors
Abstract
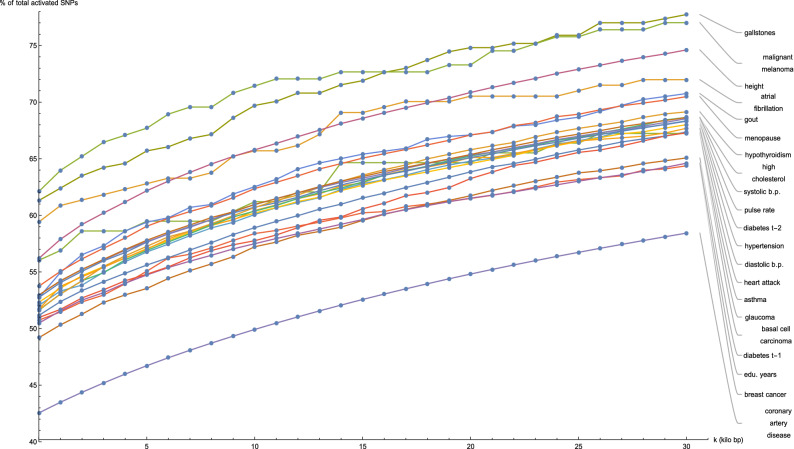
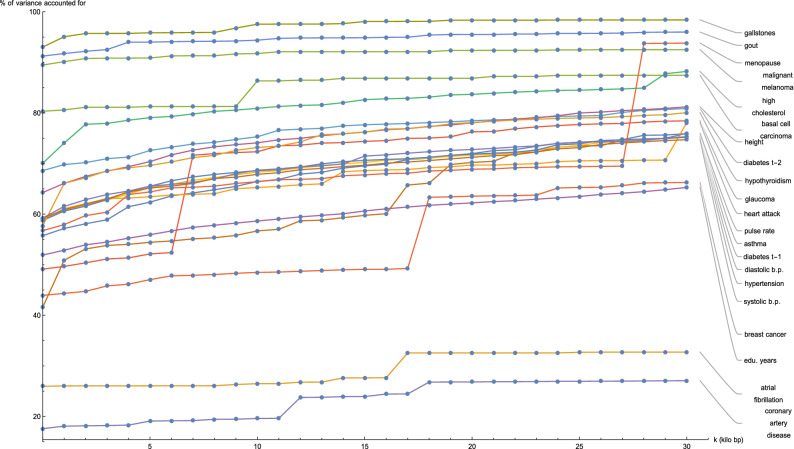
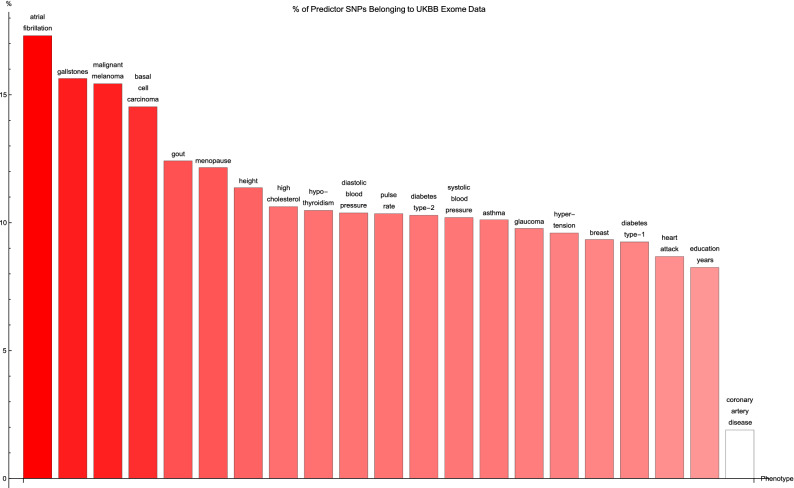
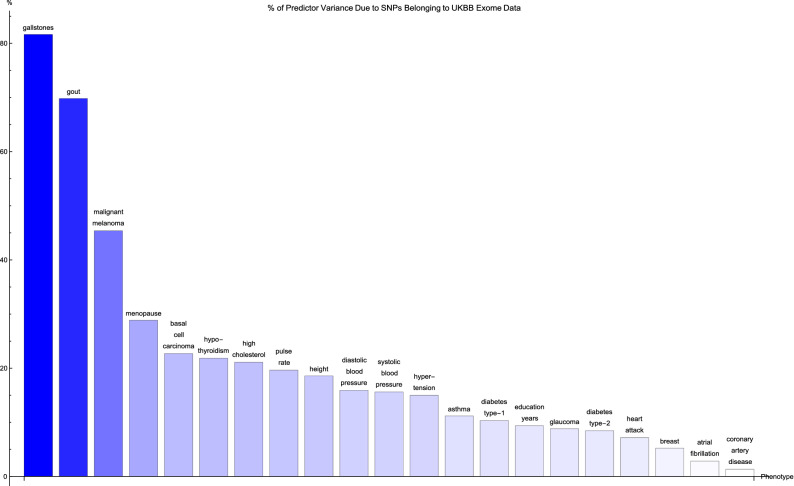
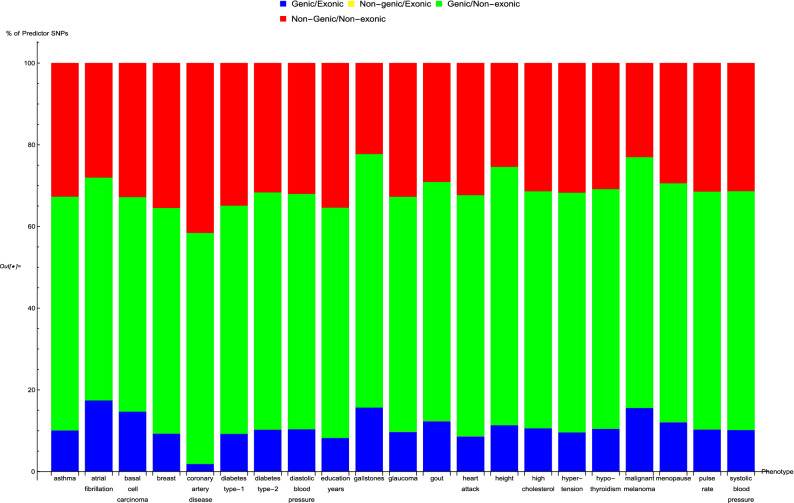
Genomic prediction of complex human traits (e.g., height, cognitive ability, bone density) and disease risks (e.g., breast cancer, diabetes, heart disease, atrial fibrillation) has advanced considerably in recent years. Using data from the UK Biobank, predictors have been constructed using penalized algorithms that favor sparsity: i.e., which use as few genetic variants as possible. We analyze the specific genetic variants (SNPs) utilized in these predictors, which can vary from dozens to as many as thirty thousand. We find that the fraction of SNPs in or near genic regions varies widely by phenotype. For the majority of disease conditions studied, a large amount of the variance is accounted for by SNPs outside of coding regions. The state of these SNPs cannot be determined from exome-sequencing data. This suggests that exome data alone will miss much of the heritability for these traits-i.e., existing PRS cannot be computed from exome data alone. We also study the fraction of SNPs and of variance that is in common between pairs of predictors. The DNA regions used in disease risk predictors so far constructed seem to be largely disjoint (with a few interesting exceptions), suggesting that individual genetic disease risks are largely uncorrelated. It seems possible in theory for an individual to be a low-risk outlier in all conditions simultaneously.
Conflict of interest statement
Stephen Hsu is a shareholder and serves on the board of directors of Genomic Prediction, Inc.. Louis Lello joined the company, becoming an employee and shareholder, during the writing and submission of this paper. Soke Yuen Yong and Timothy Raben declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
