

Identifying strategies to target the metabolic flexibility of tumours
- PMID: 32694609
- PMCID: PMC7436715
- DOI: 10.1038/s42255-020-0195-8
Identifying strategies to target the metabolic flexibility of tumours
Abstract
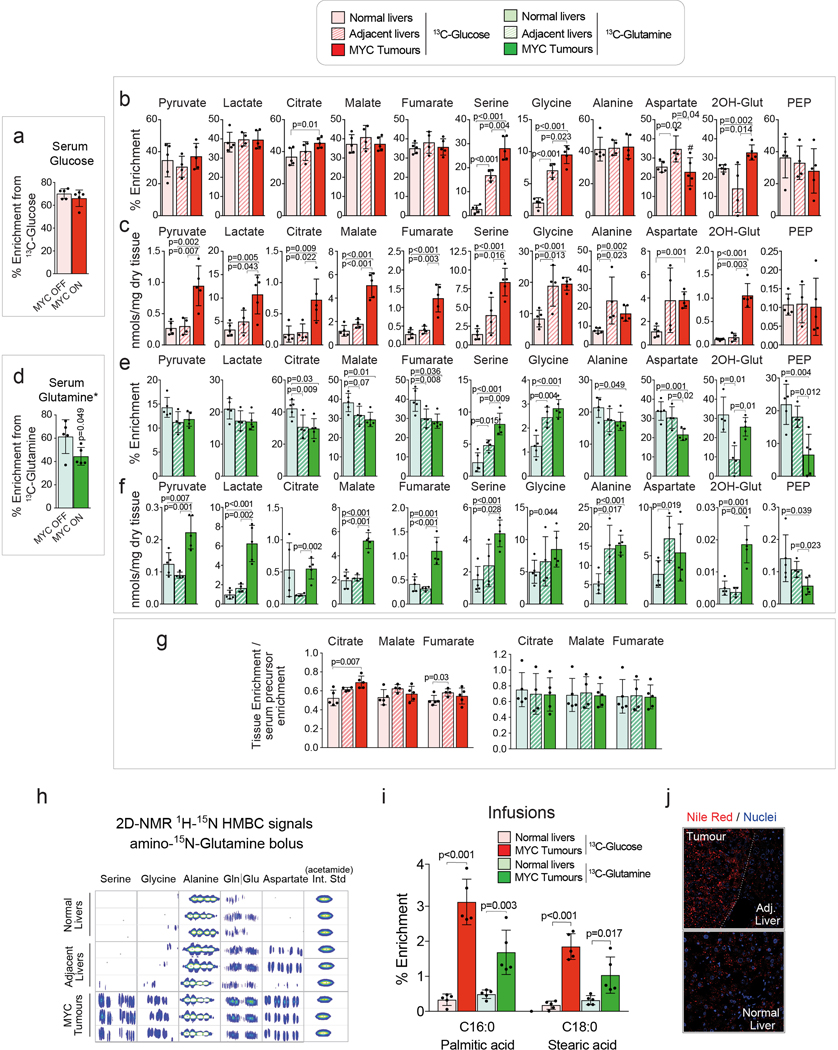
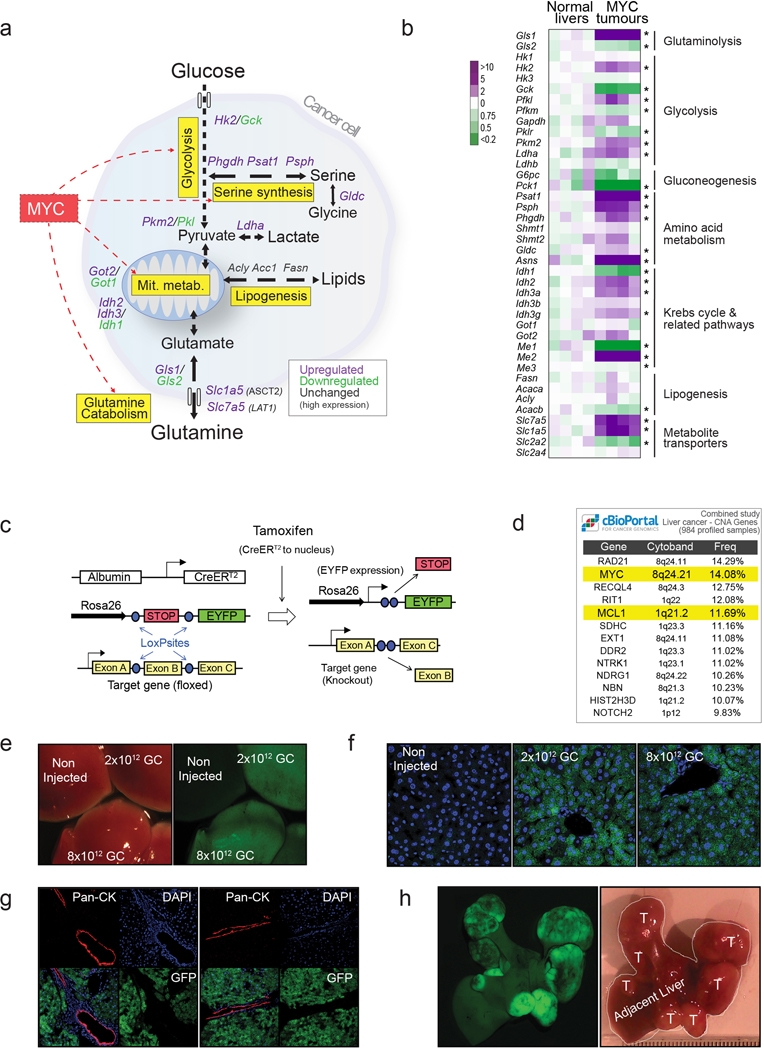
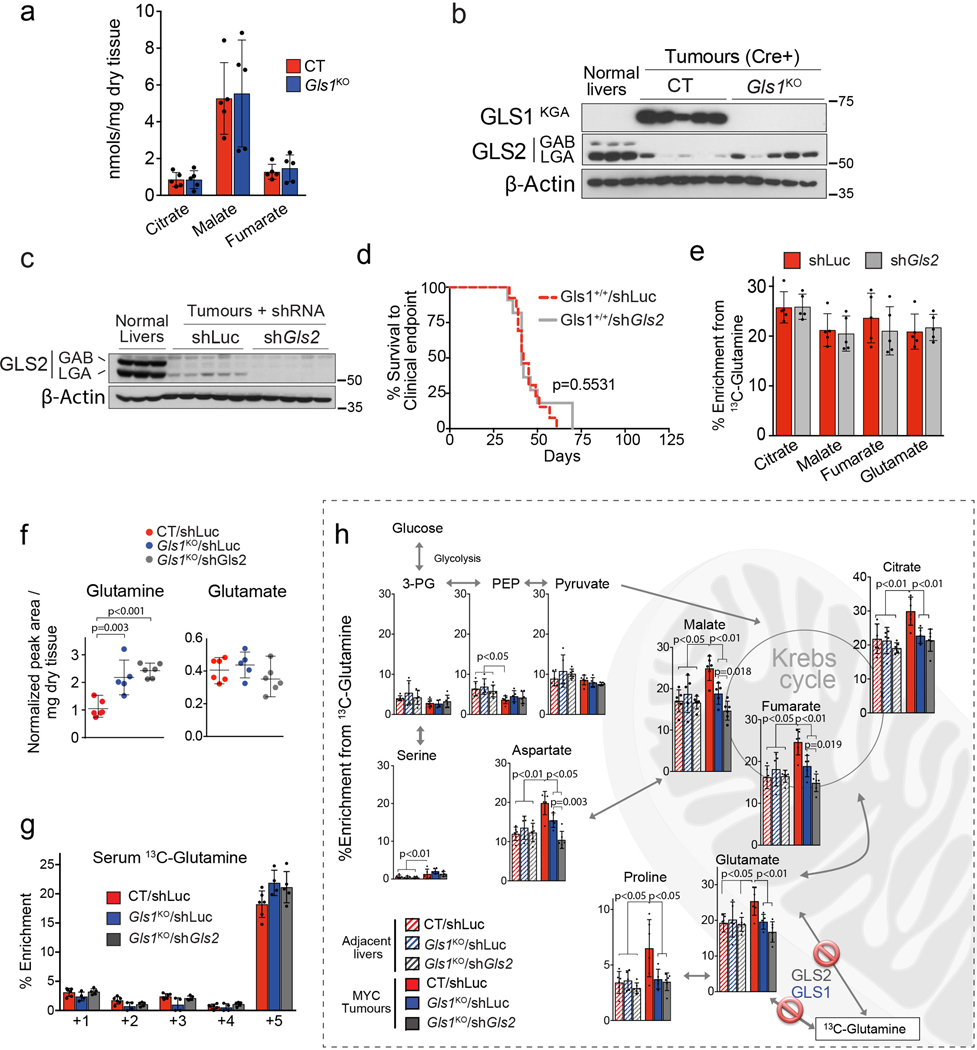
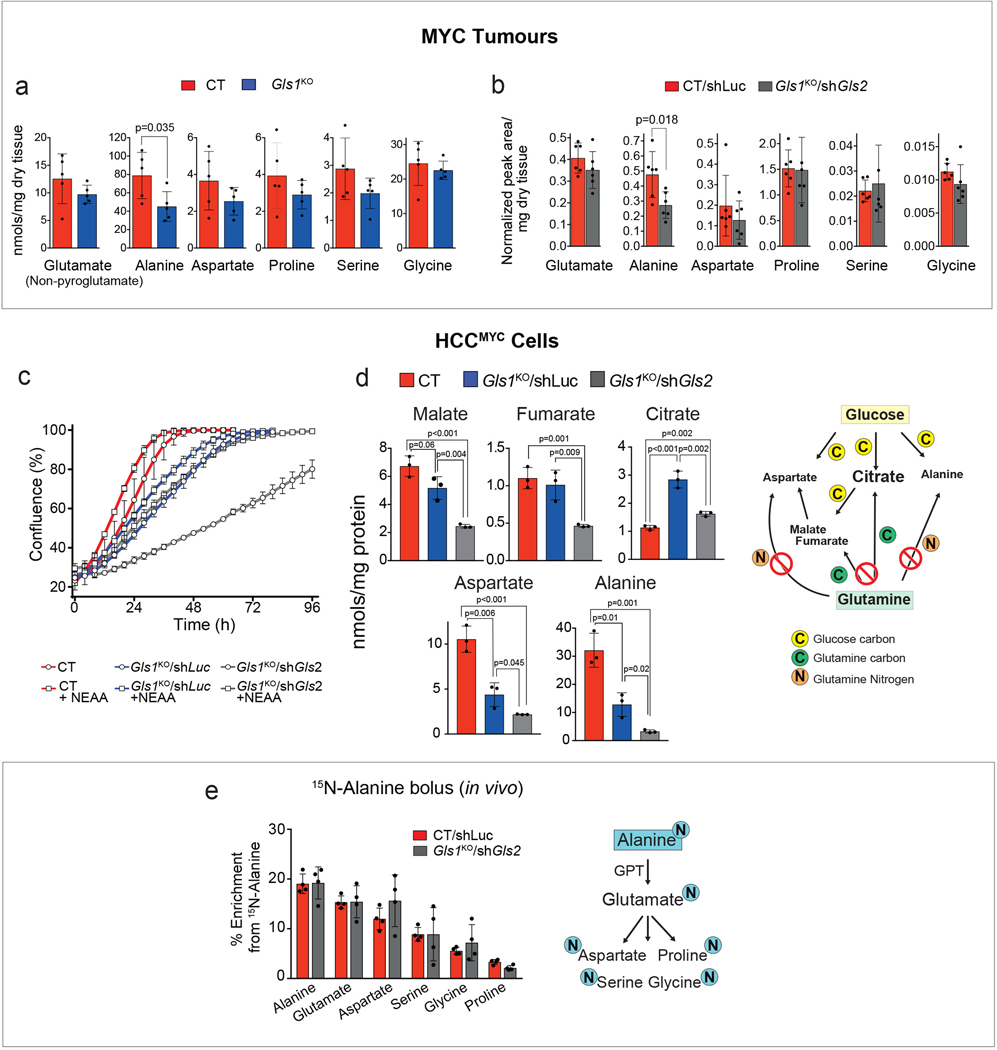
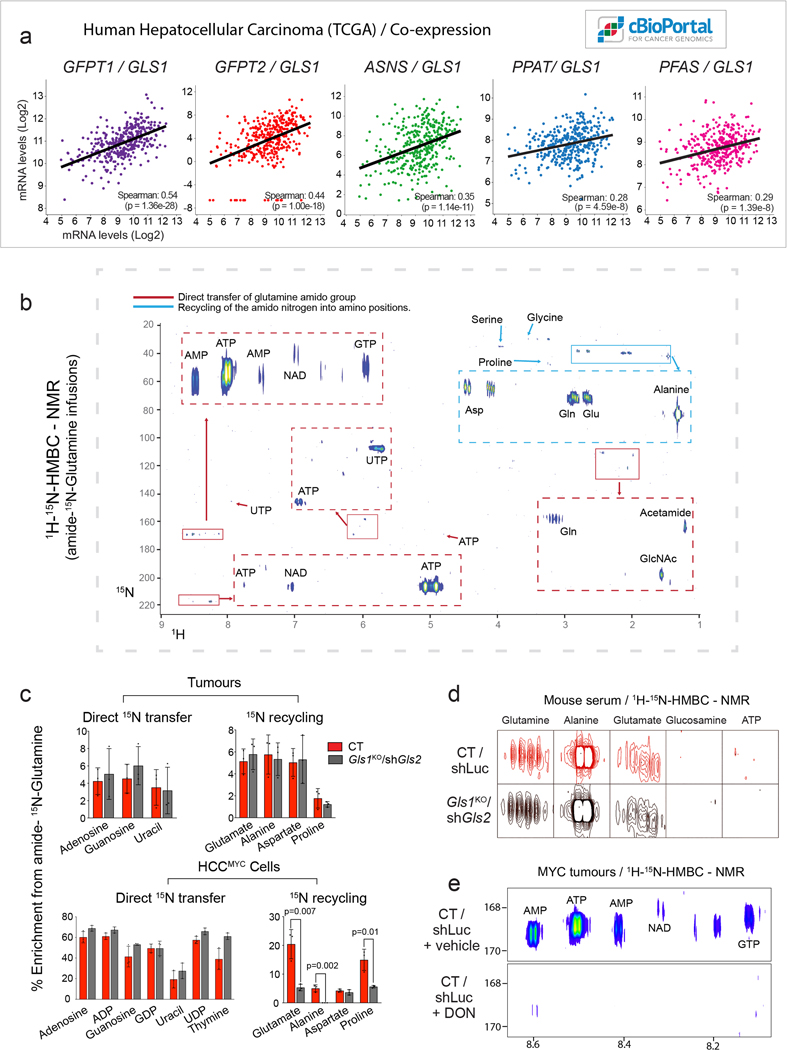
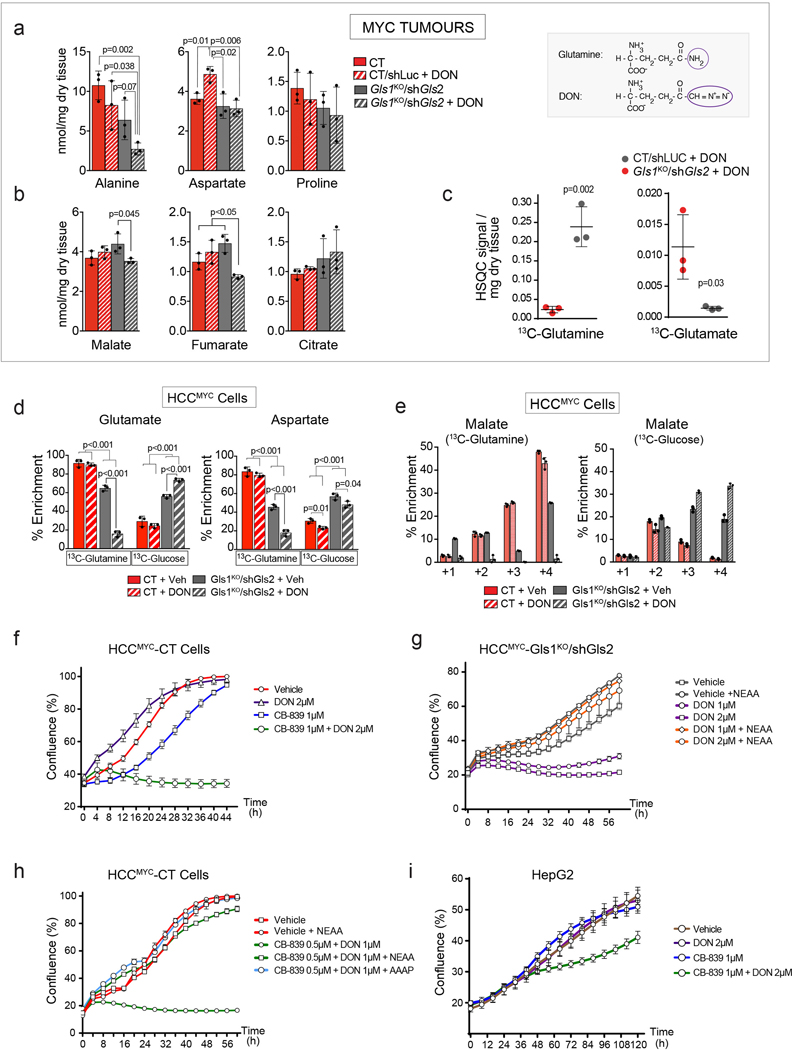
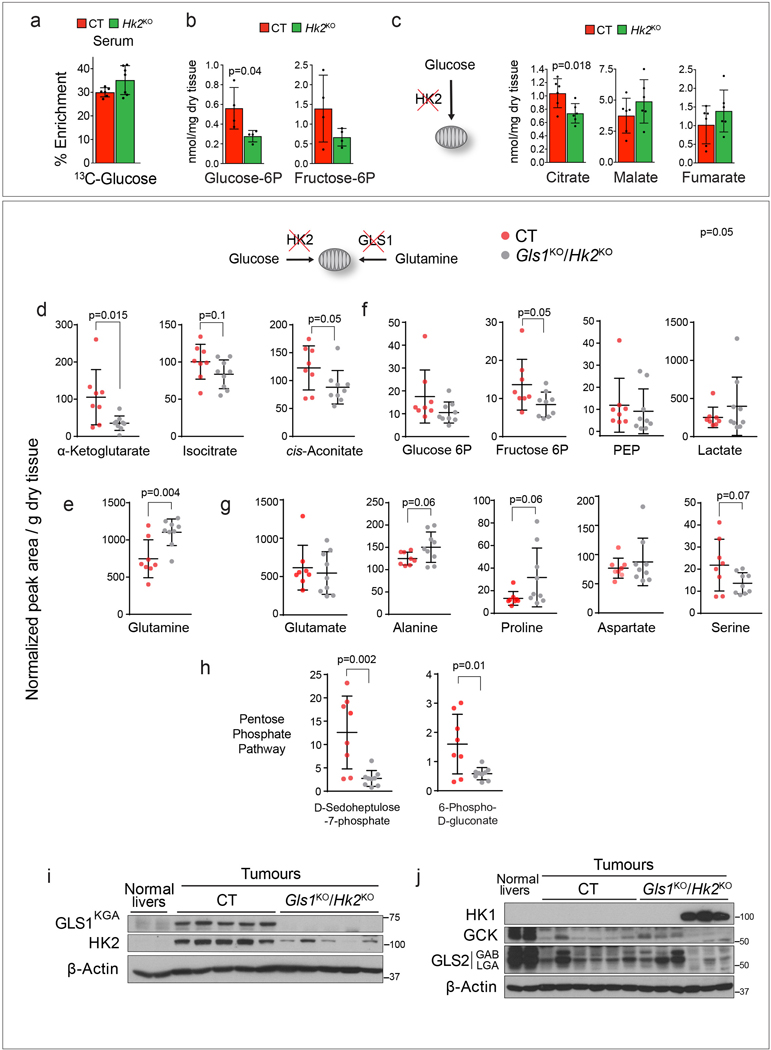
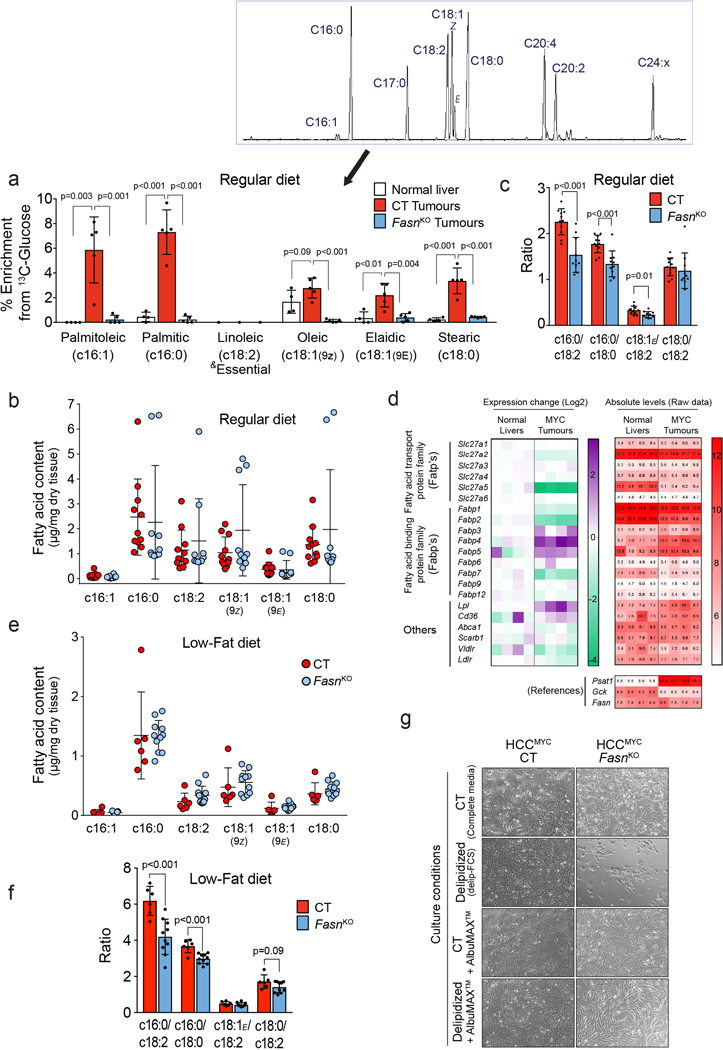
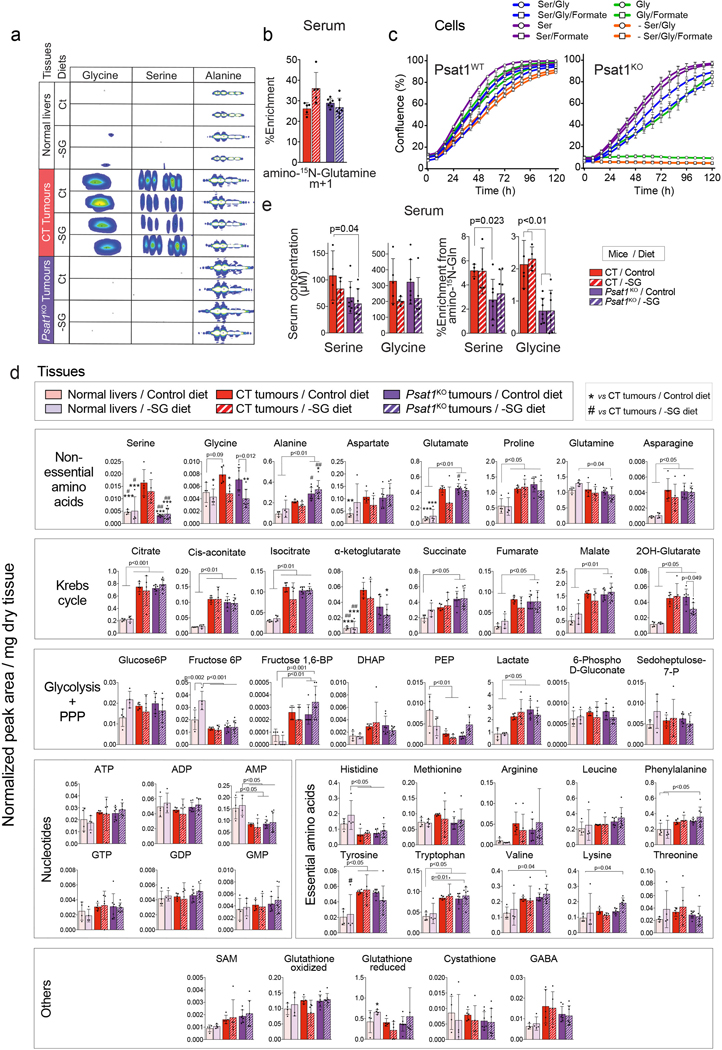
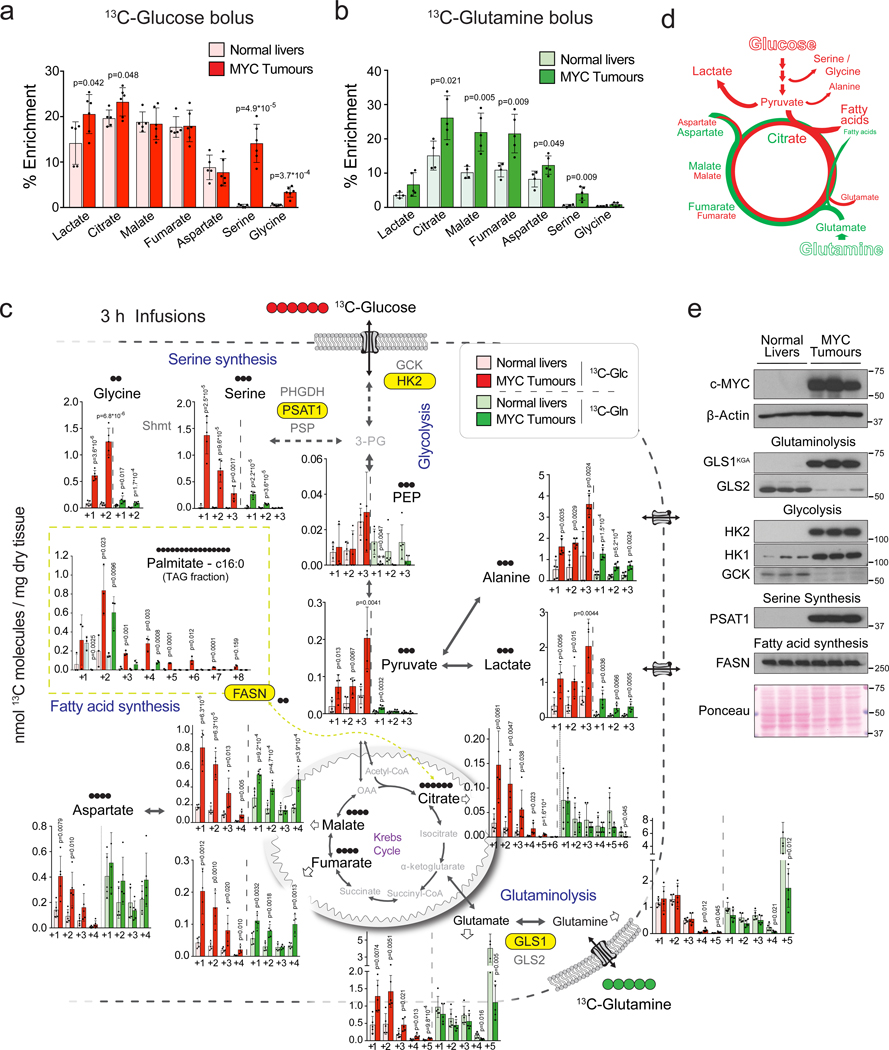
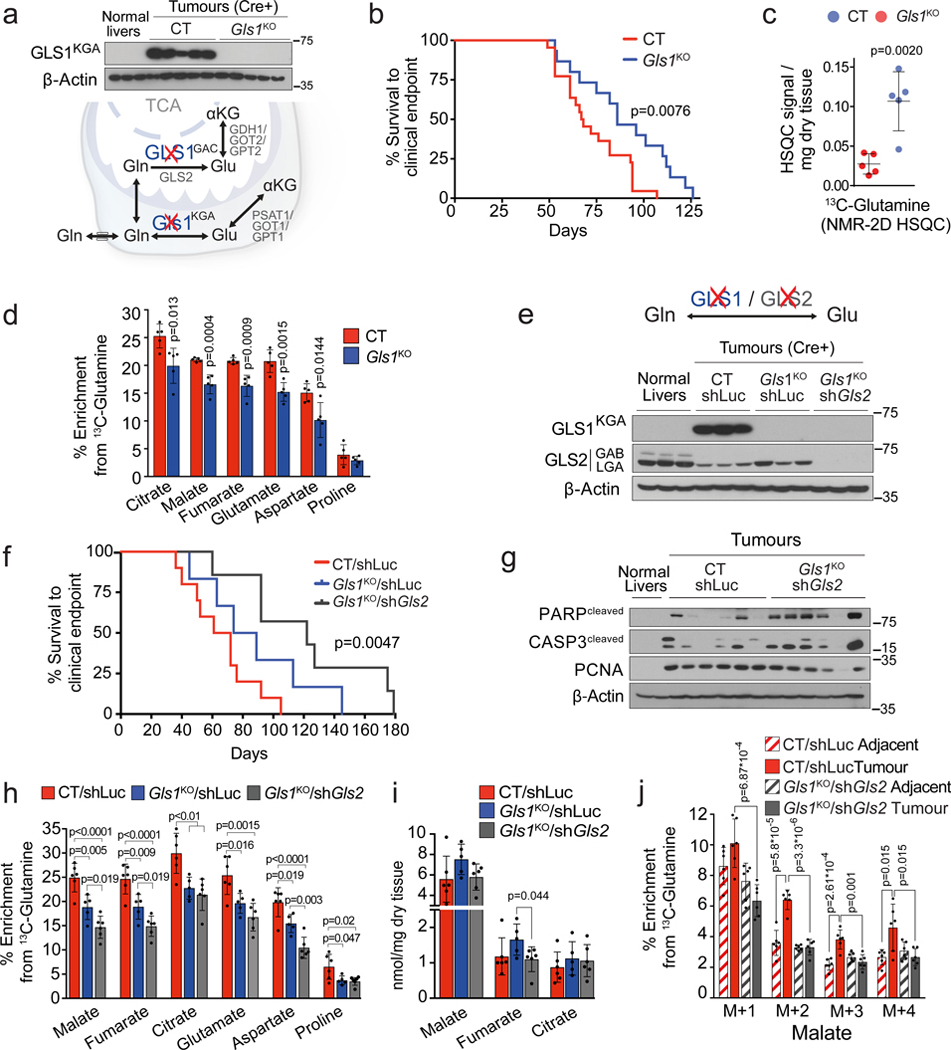
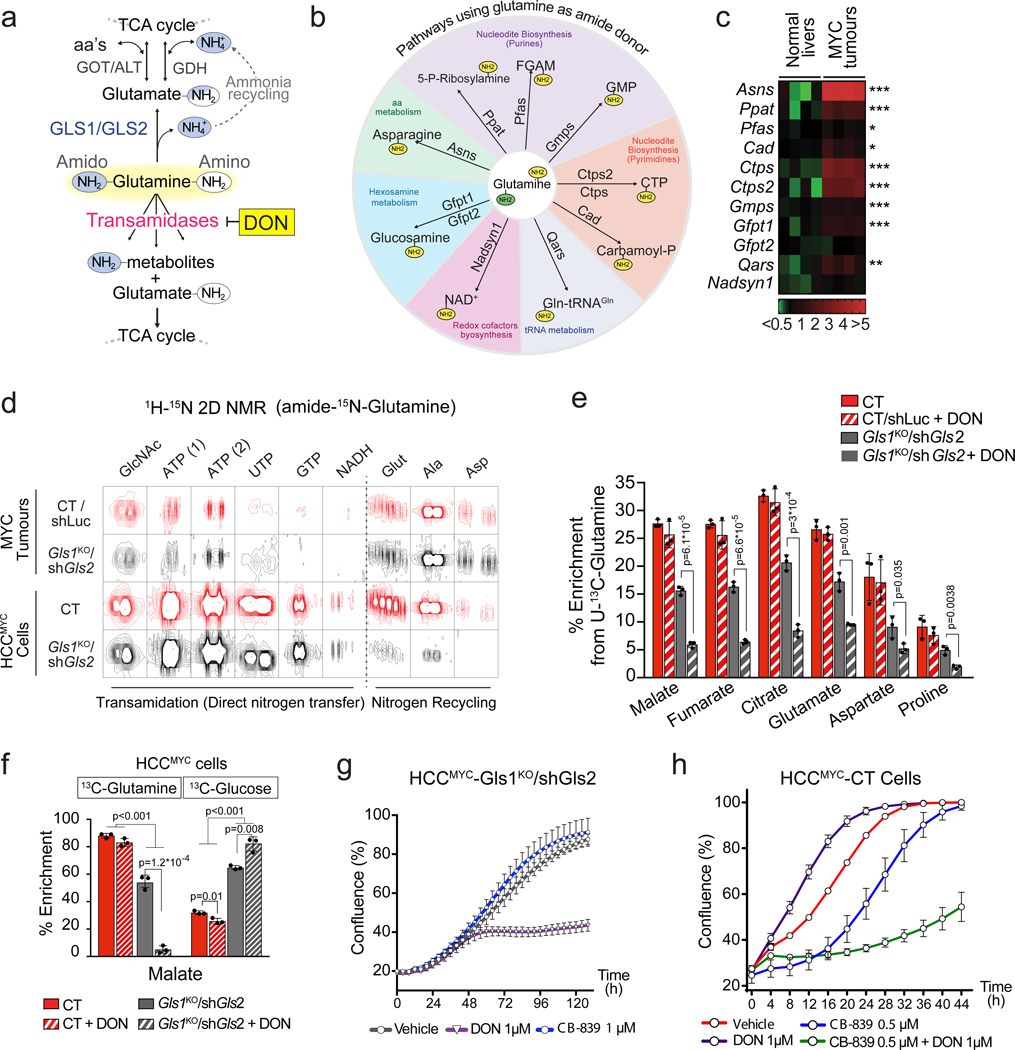
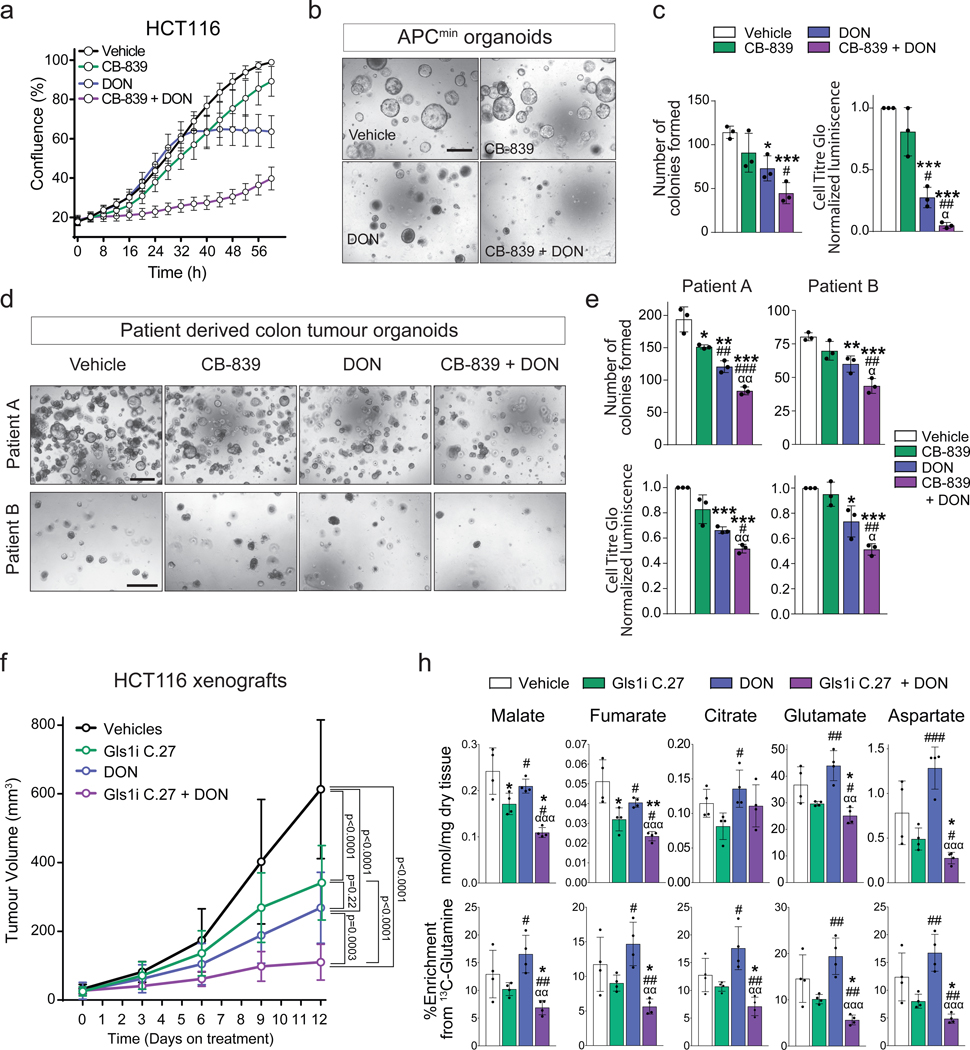
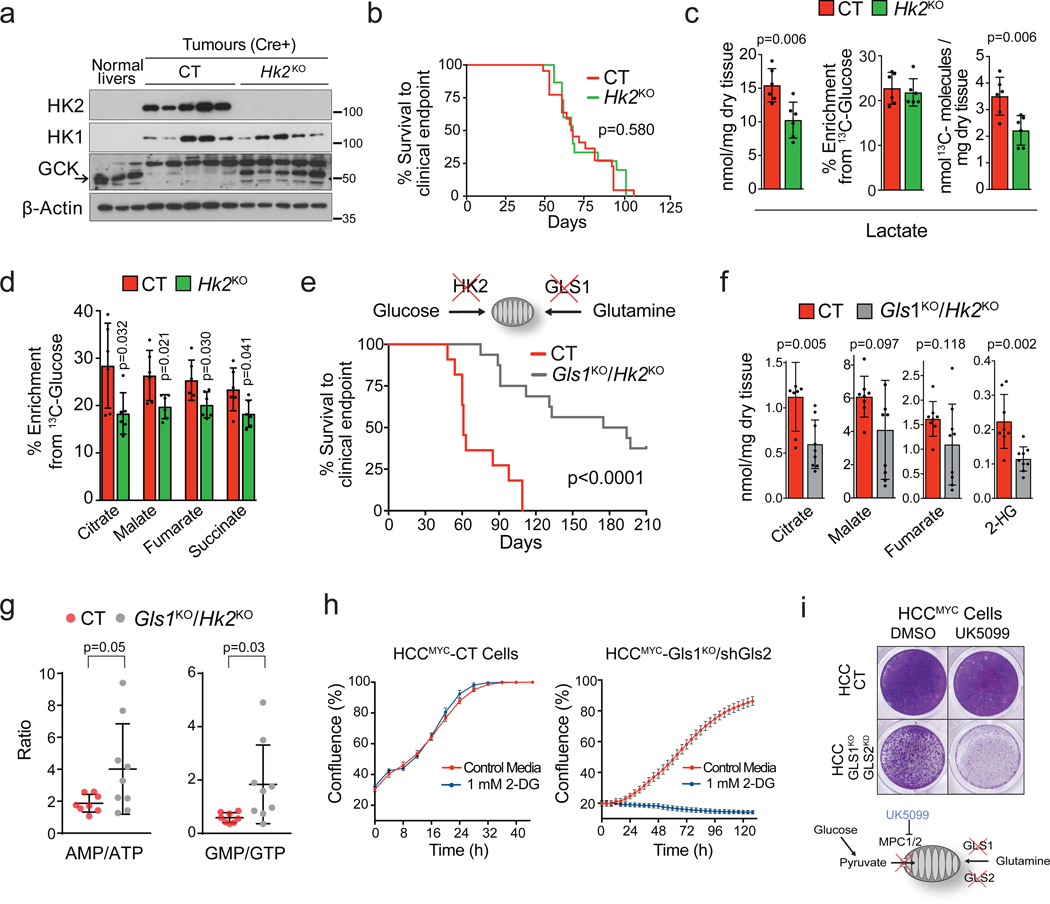
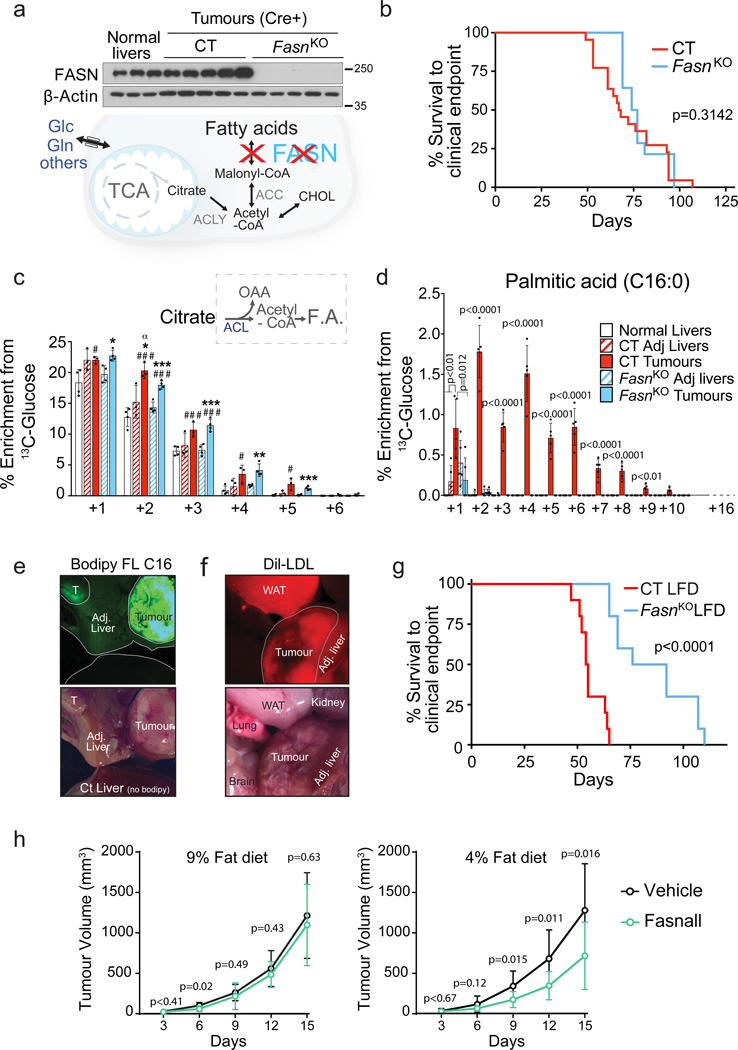
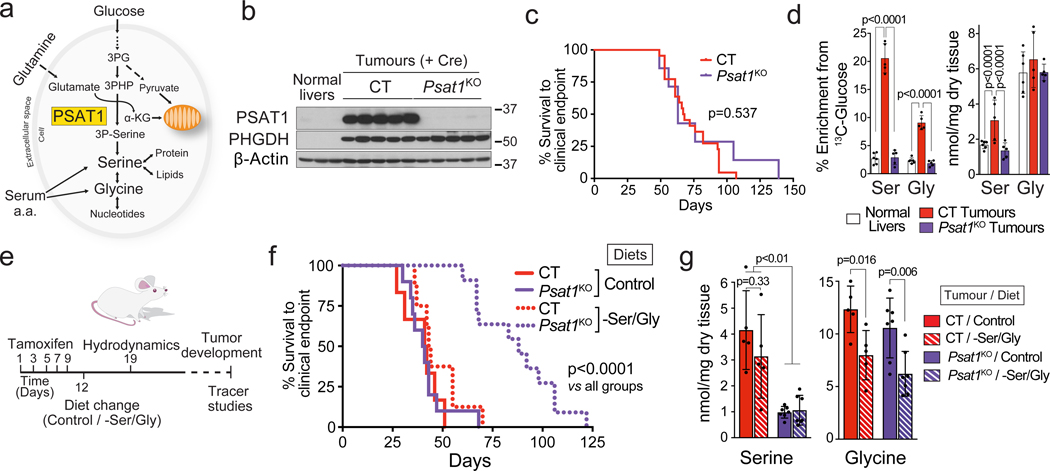
Plasticity of cancer metabolism can be a major obstacle to efficient targeting of tumour-specific metabolic vulnerabilities. Here, we identify the compensatory mechanisms following the inhibition of major pathways of central carbon metabolism in c-MYC-induced liver tumours. We find that, while inhibition of both glutaminase isoforms (Gls1 and Gls2) in tumours considerably delays tumourigenesis, glutamine catabolism continues, owing to the action of amidotransferases. Synergistic inhibition of both glutaminases and compensatory amidotransferases is required to block glutamine catabolism and proliferation of mouse and human tumour cells in vitro and in vivo. Gls1 deletion is also compensated for by glycolysis. Thus, co-inhibition of Gls1 and hexokinase 2 significantly affects Krebs cycle activity and tumour formation. Finally, the inhibition of biosynthesis of either serine (Psat1-KO) or fatty acid (Fasn-KO) is compensated for by uptake of circulating nutrients, and dietary restriction of both serine and glycine or fatty acids synergistically suppresses tumourigenesis. These results highlight the high flexibility of tumour metabolism and demonstrate that either pharmacological or dietary targeting of metabolic compensatory mechanisms can improve therapeutic outcomes.
Figures
References
Methods-only References
Publication types
MeSH terms
Substances
Grants and funding
- IK6 BX004602/BX/BLRD VA/United States
- P30 DK026743/DK/NIDDK NIH HHS/United States
- MC_U117533887/MRC_/Medical Research Council/United Kingdom
- R01 CA206167/CA/NCI NIH HHS/United States
- FC001223/WT_/Wellcome Trust/United Kingdom
- R21 CA198490/CA/NCI NIH HHS/United States
- I01 BX000733/BX/BLRD VA/United States
- R01 AG016927/AG/NIA NIH HHS/United States
- R01 CA090764/CA/NCI NIH HHS/United States
- FC001223/CRUK_/Cancer Research UK/United Kingdom
- R01 CA136606/CA/NCI NIH HHS/United States
- FC001223/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
