

Systematic mapping of genetic interactions for de novo fatty acid synthesis identifies C12orf49 as a regulator of lipid metabolism
- PMID: 32694731
- PMCID: PMC7566881
- DOI: 10.1038/s42255-020-0211-z
Systematic mapping of genetic interactions for de novo fatty acid synthesis identifies C12orf49 as a regulator of lipid metabolism
Abstract
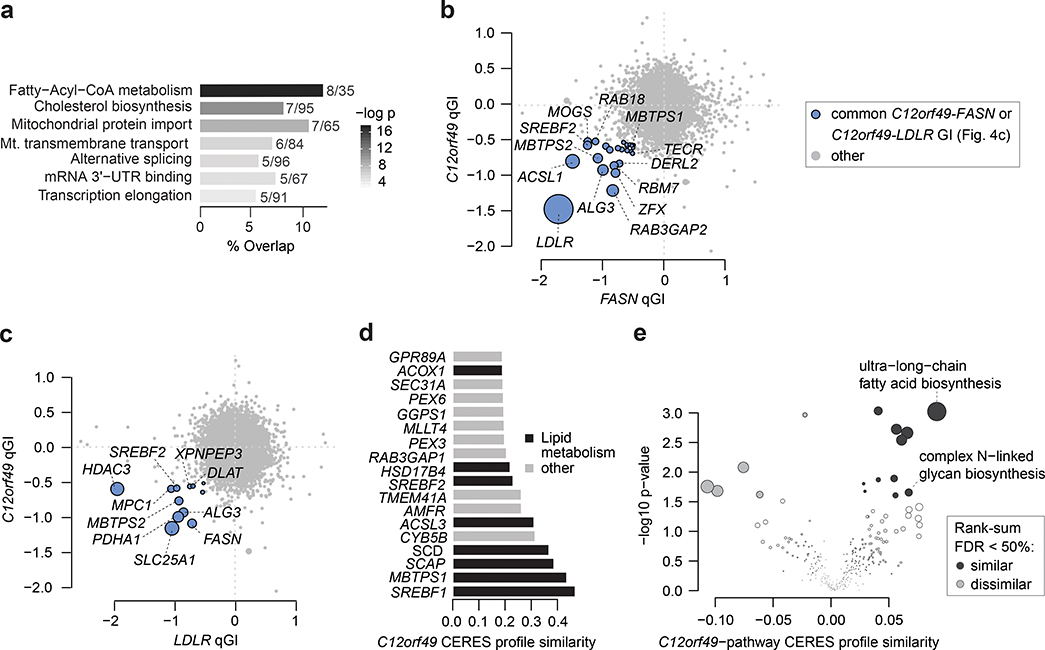
The de novo synthesis of fatty acids has emerged as a therapeutic target for various diseases, including cancer. Because cancer cells are intrinsically buffered to combat metabolic stress, it is important to understand how cells may adapt to the loss of de novo fatty acid biosynthesis. Here, we use pooled genome-wide CRISPR screens to systematically map genetic interactions (GIs) in human HAP1 cells carrying a loss-of-function mutation in fatty acid synthase (FASN), whose product catalyses the formation of long-chain fatty acids. FASN-mutant cells show a strong dependence on lipid uptake that is reflected in negative GIs with genes involved in the LDL receptor pathway, vesicle trafficking and protein glycosylation. Further support for these functional relationships is derived from additional GI screens in query cell lines deficient in other genes involved in lipid metabolism, including LDLR, SREBF1, SREBF2 and ACACA. Our GI profiles also identify a potential role for the previously uncharacterized gene C12orf49 (which we call LUR1) in regulation of exogenous lipid uptake through modulation of SREBF2 signalling in response to lipid starvation. Overall, our data highlight the genetic determinants underlying the cellular adaptation associated with loss of de novo fatty acid synthesis and demonstrate the power of systematic GI mapping for uncovering metabolic buffering mechanisms in human cells.
Conflict of interest statement
COMPETING INTERESTS STATEMENT
J.M., B.A. and C.B are shareholders in Northern Biologics. J.M. is a shareholder in Pionyr Immunotherapeutics, is acting CSO and shareholder in Empirica Therapeutics, and is an SAB member and shareholder of Aelian Biotechnology. C.B. is an SAB member of Yumanity Therapeutics. The authors declare no competing interest.
Figures












References
-
- Imoto M Chemistry and biology for the small molecules targeting characteristics of cancer cells. Biosci. Biotechnol. Biochem. 83, 1–10 (2018). - PubMed
-
- Menendez JA & Lupu R Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 21, 1001–1016 (2017). - PubMed
-
- Garber K Cancer anabolic metabolism inhibitors move into clinic. Nat. Biotechnol. 34, 794–795 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
