

Accelerating Therapeutics for Opportunities in Medicine: A Paradigm Shift in Drug Discovery
- PMID: 32694991
- PMCID: PMC7339658
- DOI: 10.3389/fphar.2020.00770
Accelerating Therapeutics for Opportunities in Medicine: A Paradigm Shift in Drug Discovery
Abstract
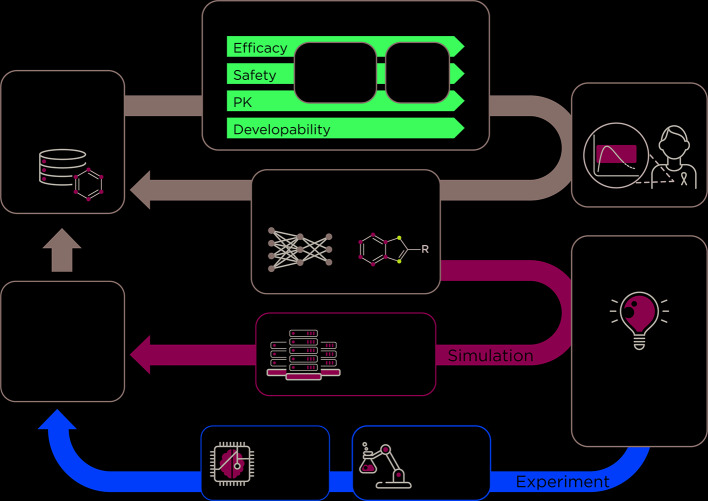
Conventional drug discovery is long and costly, and suffers from high attrition rates, often leaving patients with limited or expensive treatment options. Recognizing the overwhelming need to accelerate this process and increase success, the ATOM consortium was formed by government, industry, and academic partners in October 2017. ATOM applies a team science and open-source approach to foster a paradigm shift in drug discovery. ATOM is developing and validating a precompetitive, preclinical, small molecule drug discovery platform that simultaneously optimizes pharmacokinetics, toxicity, protein-ligand interactions, systems-level models, molecular design, and novel compound generation. To achieve this, the ATOM Modeling Pipeline (AMPL) has been developed to enable advanced and emerging machine learning (ML) approaches to build models from diverse historical drug discovery data. This modular pipeline has been designed to couple with a generative algorithm that optimizes multiple parameters necessary for drug discovery. ATOM's approach is to consider the full pharmacology and therapeutic window of the drug concurrently, through computationally-driven design, thereby reducing the number of molecules that are selected for experimental validation. Here, we discuss the role of collaborative efforts such as consortia and public-private partnerships in accelerating cross disciplinary innovation and the development of open-source tools for drug discovery.
Keywords: artificial intelligence; data science; drug discovery and development; in silico modeling; machine learning.
Copyright © 2020 Hinkson, Madej and Stahlberg.
Figures
References
-
- Berthold M. R. E. A. (2008). “KNIME: The Konstanz Information Miner,” in Data Analysis, Machine Learning and Applications. Studies in Classification, Data Analysis, and Knowledge Organization. Eds. Preisach C. B. H., Schmidt-Thieme L., Decker R. (Berlin, Heidelberg: Springer; ).
Grants and funding
LinkOut - more resources
Full Text Sources