

Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus
- PMID: 32697968
- PMCID: PMC7332439
- DOI: 10.1016/j.cell.2020.06.043
Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus
Abstract
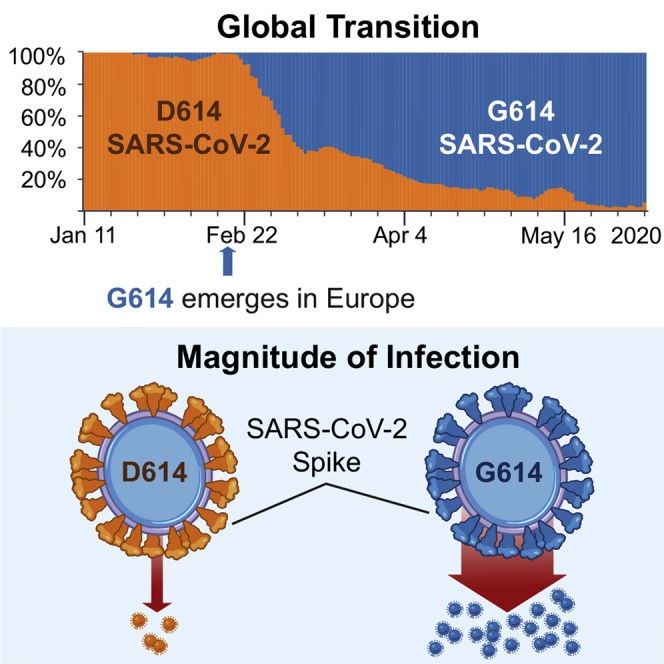
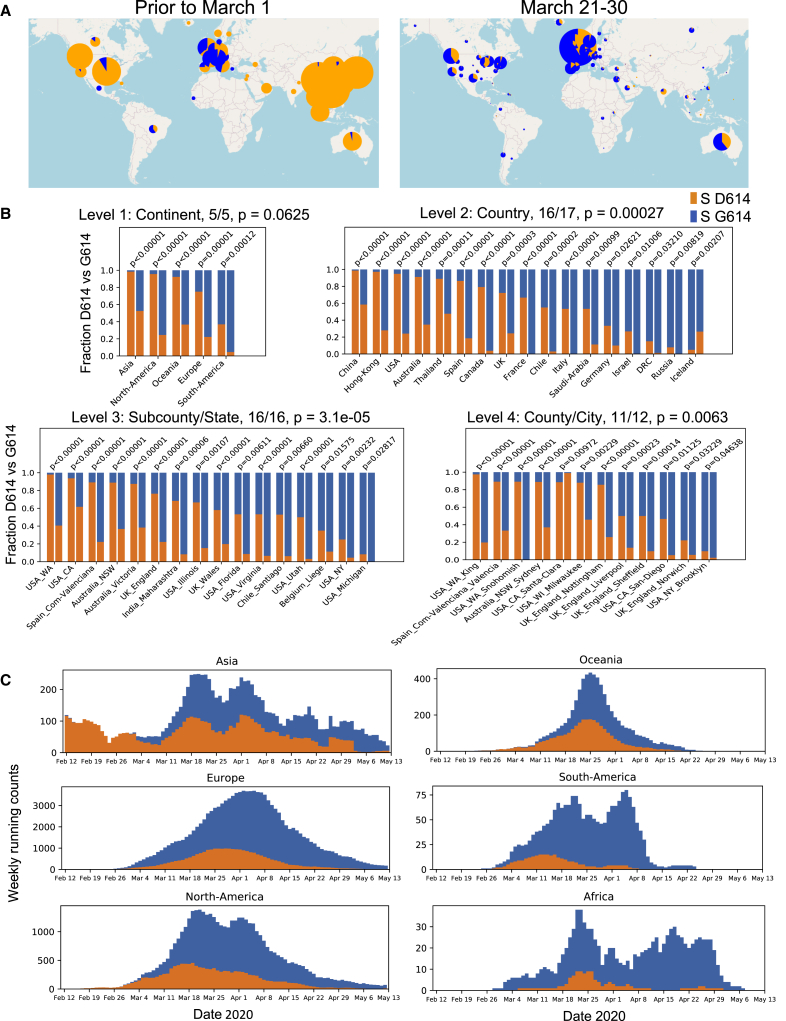
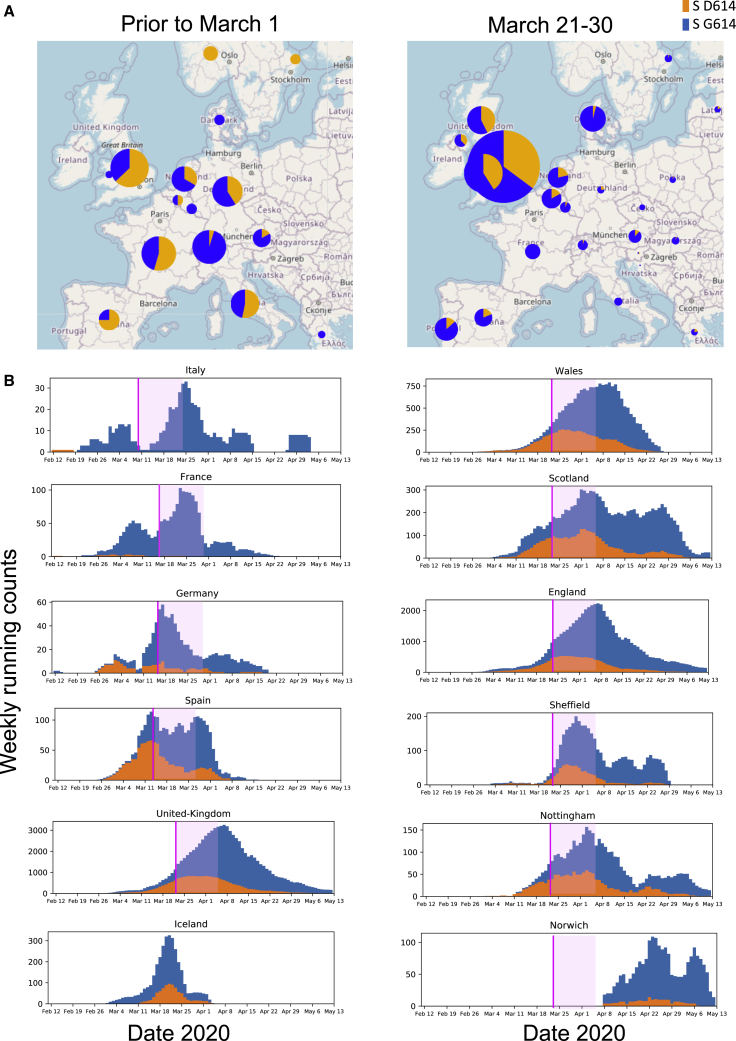
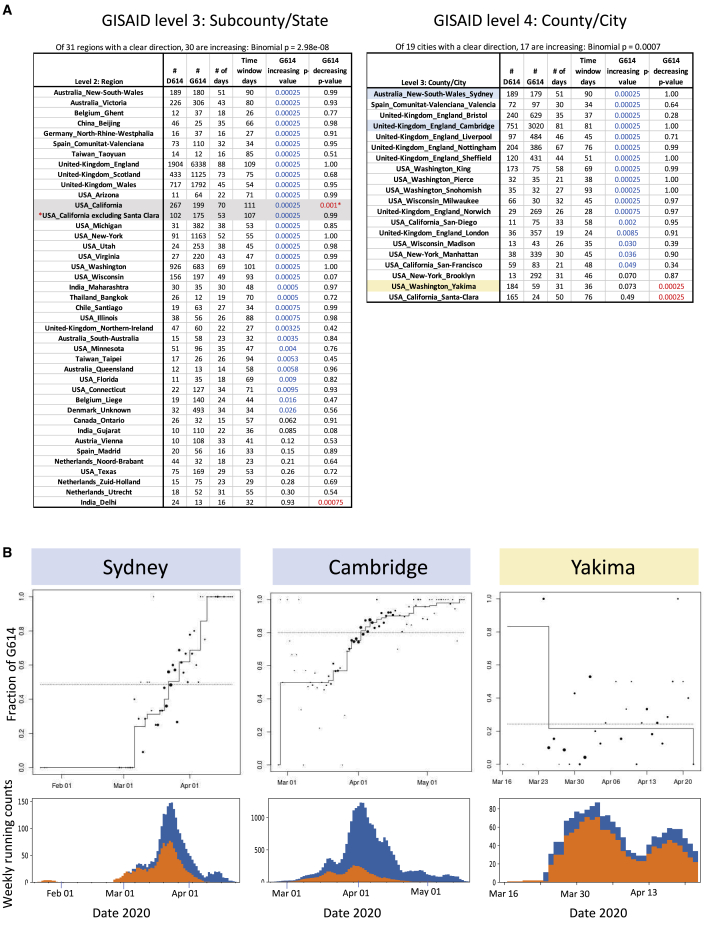
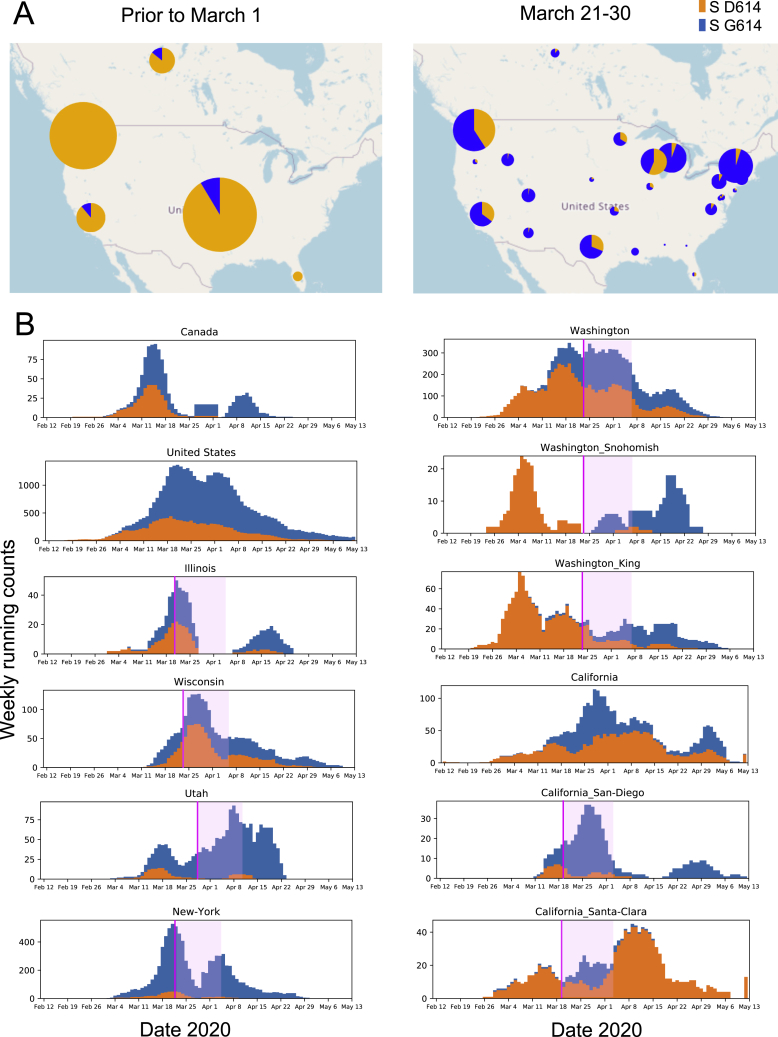
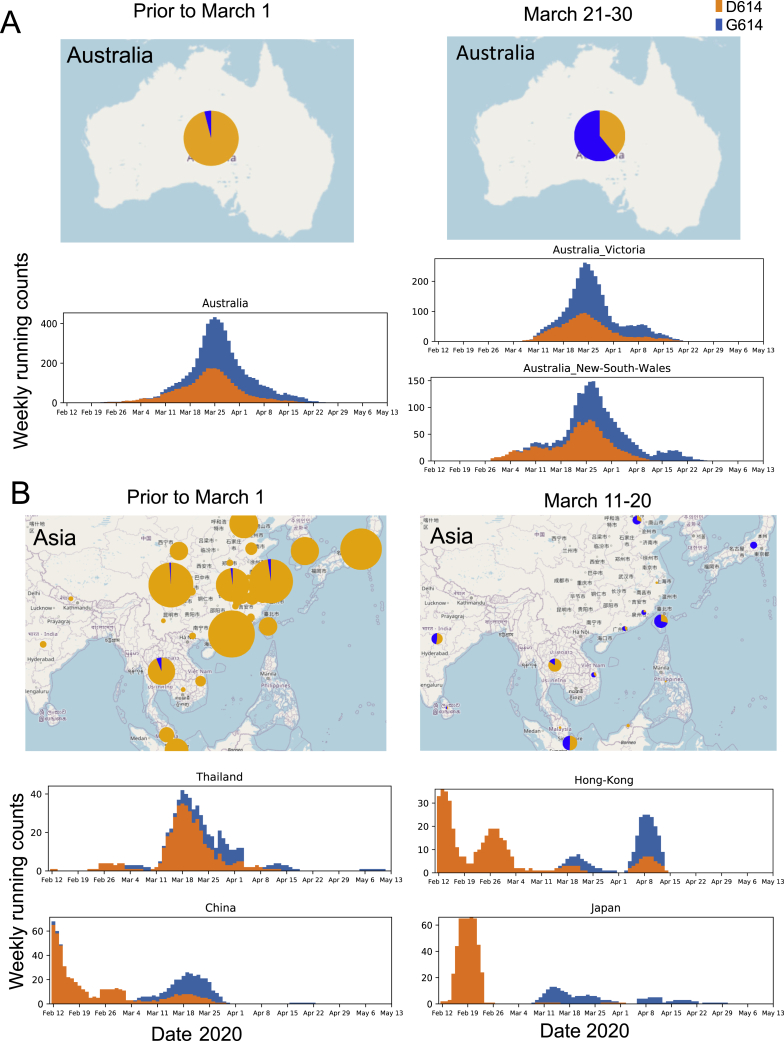
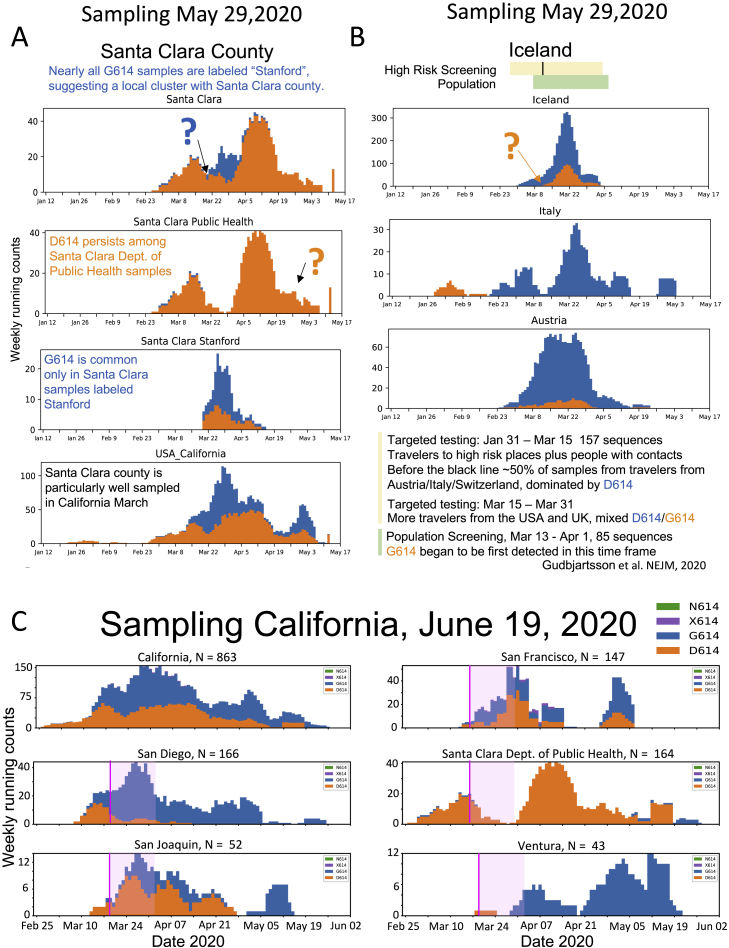
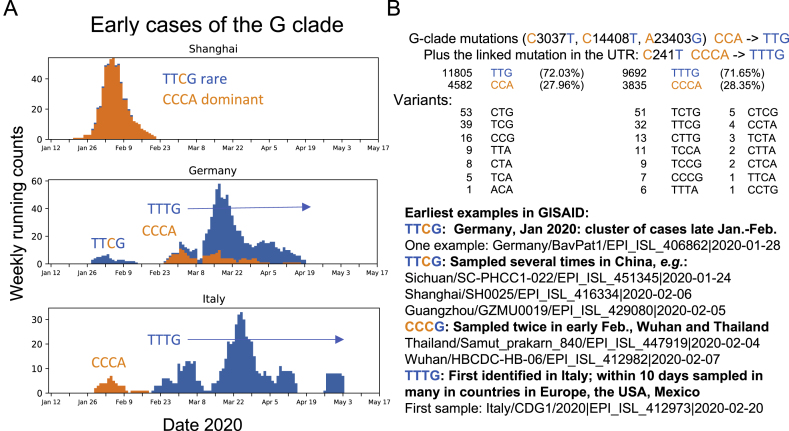
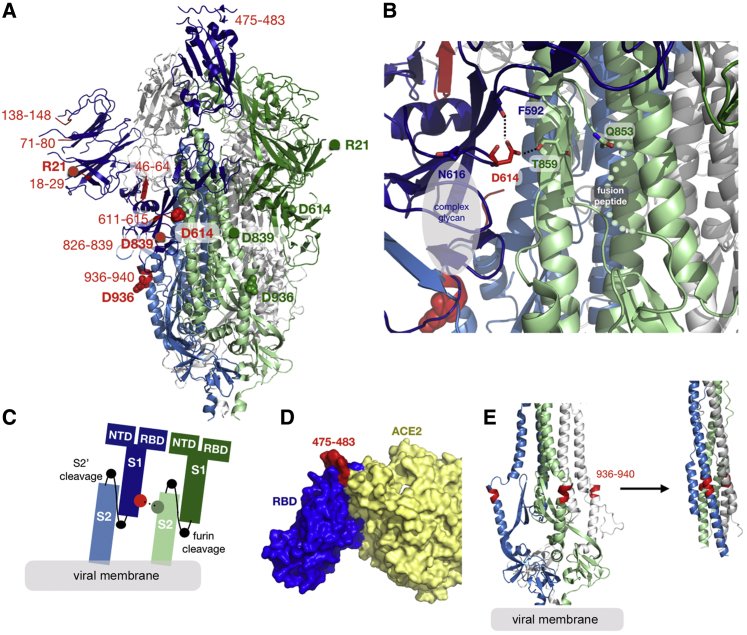
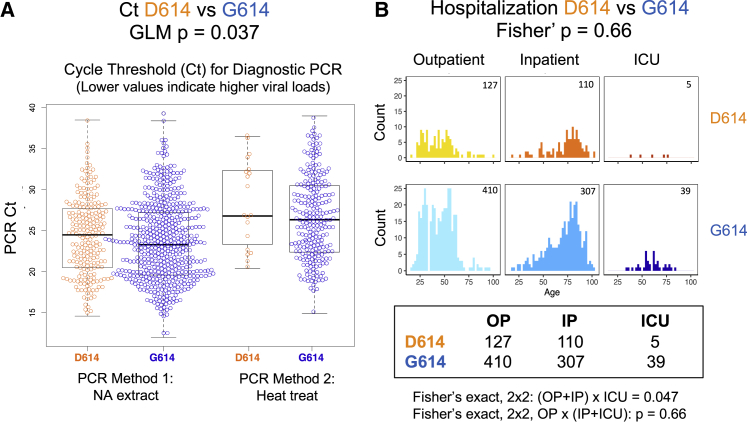
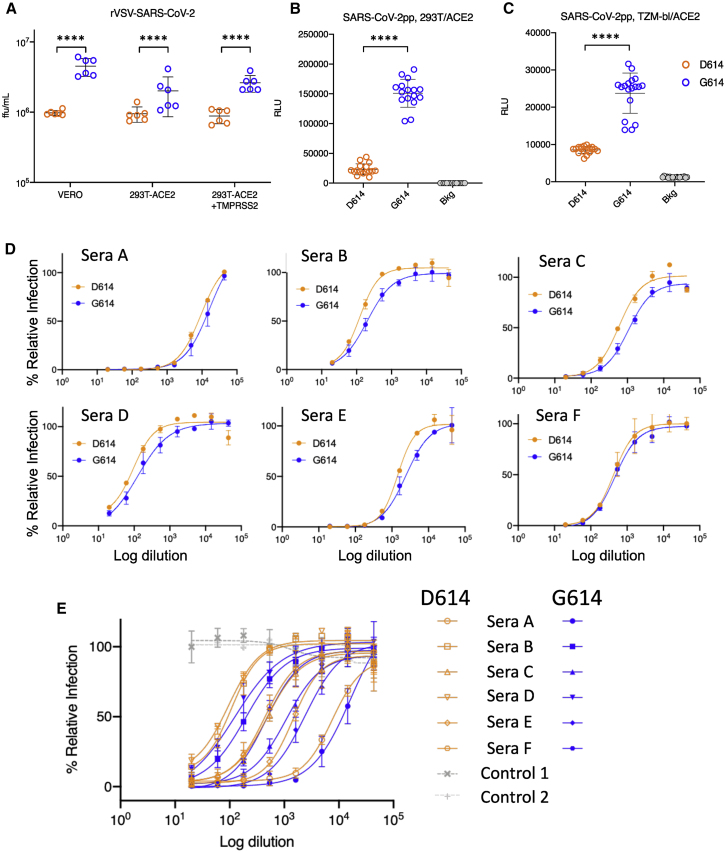
A SARS-CoV-2 variant carrying the Spike protein amino acid change D614G has become the most prevalent form in the global pandemic. Dynamic tracking of variant frequencies revealed a recurrent pattern of G614 increase at multiple geographic levels: national, regional, and municipal. The shift occurred even in local epidemics where the original D614 form was well established prior to introduction of the G614 variant. The consistency of this pattern was highly statistically significant, suggesting that the G614 variant may have a fitness advantage. We found that the G614 variant grows to a higher titer as pseudotyped virions. In infected individuals, G614 is associated with lower RT-PCR cycle thresholds, suggestive of higher upper respiratory tract viral loads, but not with increased disease severity. These findings illuminate changes important for a mechanistic understanding of the virus and support continuing surveillance of Spike mutations to aid with development of immunological interventions.
Keywords: COVID-19; PCR cycle threshold; SARS-CoV-2; Spike; antibody; diversity; evolution; infectivity; neutralization; pseudovirus.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
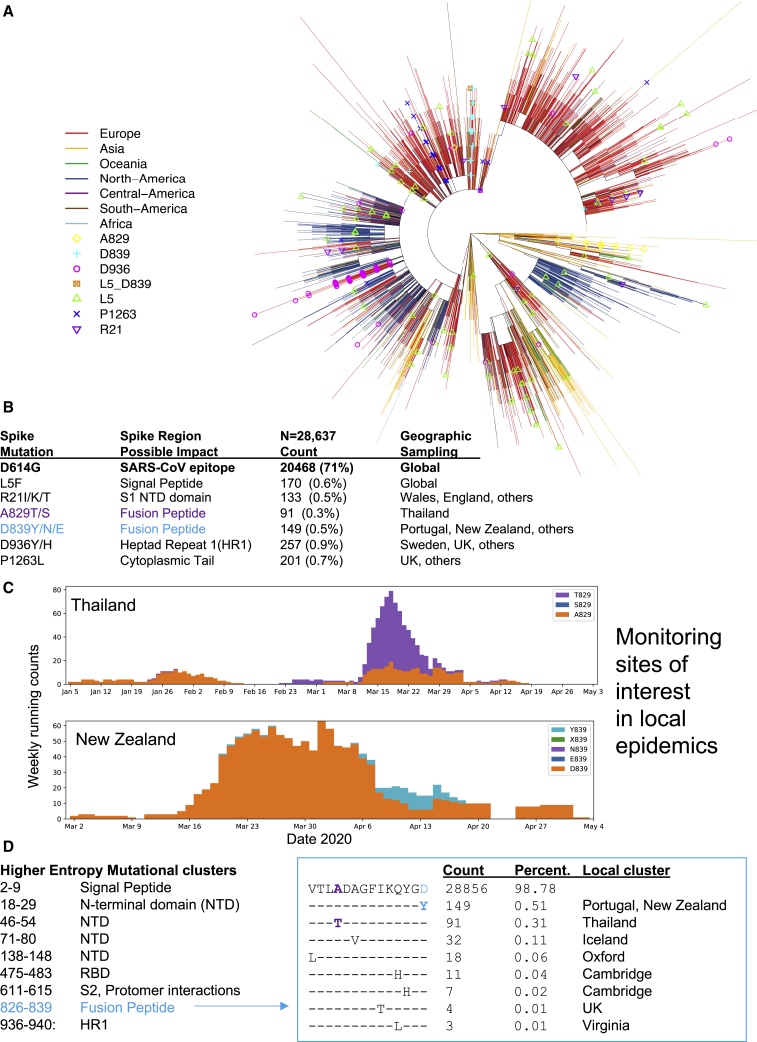
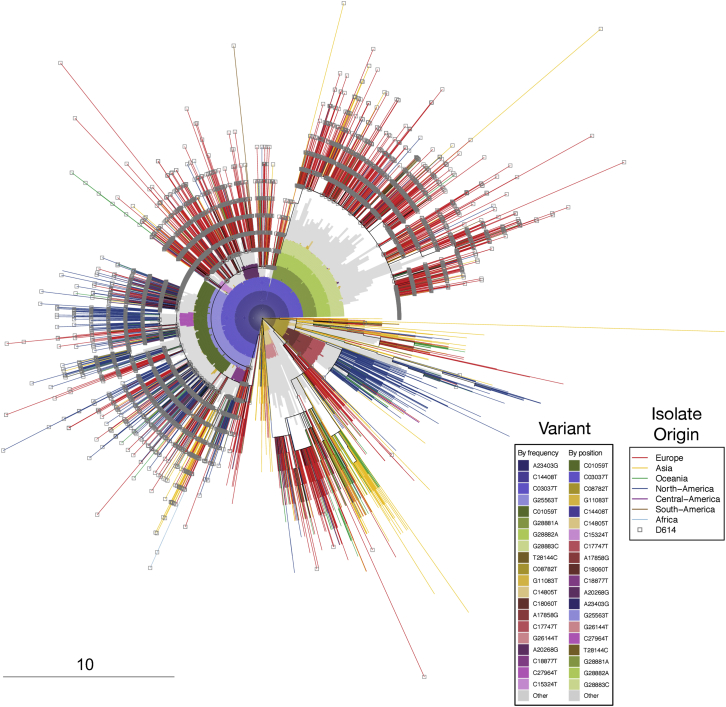
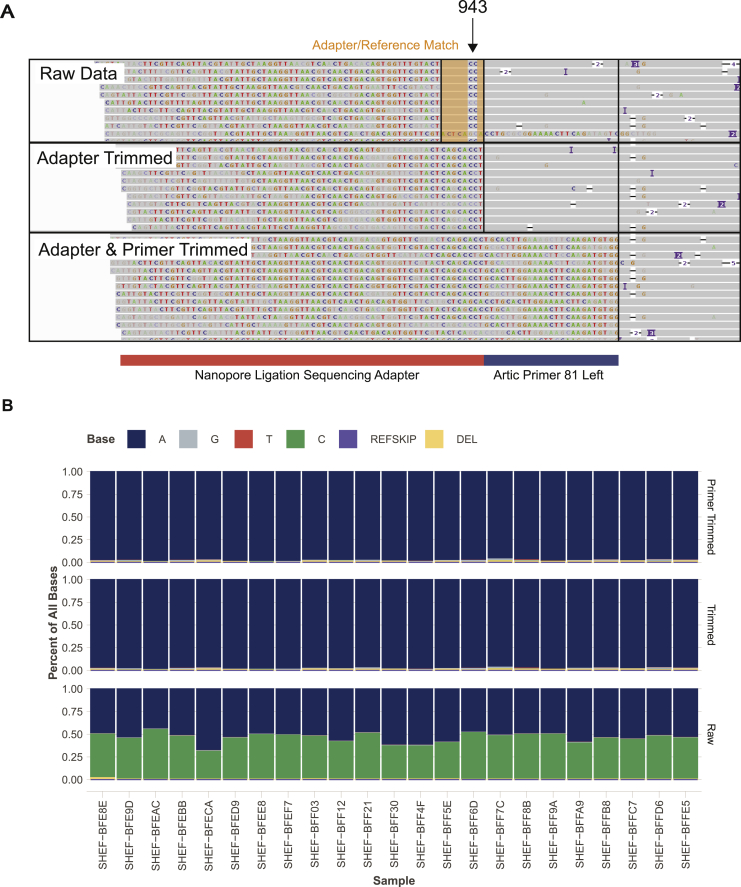
Comment in
-
Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear.Cell. 2020 Aug 20;182(4):794-795. doi: 10.1016/j.cell.2020.06.040. Epub 2020 Jul 3. Cell. 2020. PMID: 32697970 Free PMC article.
-
Emergence of a new SARS-CoV-2 variant in the UK.J Infect. 2021 Apr;82(4):e27-e28. doi: 10.1016/j.jinf.2020.12.024. Epub 2020 Dec 28. J Infect. 2021. PMID: 33383088 Free PMC article. No abstract available.
References
-
- Asmal M., Hellmann I., Liu W., Keele B.F., Perelson A.S., Bhattacharya T., Gnanakaran S., Daniels M., Haynes B.F., Korber B.T. A signature in HIV-1 envelope leader peptide associated with transition from acute to chronic infection impacts envelope processing and infectivity. PLoS ONE. 2011;6:e23673. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
