

Controlling the SARS-CoV-2 spike glycoprotein conformation
- PMID: 32699321
- PMCID: PMC8581954
- DOI: 10.1038/s41594-020-0479-4
Controlling the SARS-CoV-2 spike glycoprotein conformation
Abstract
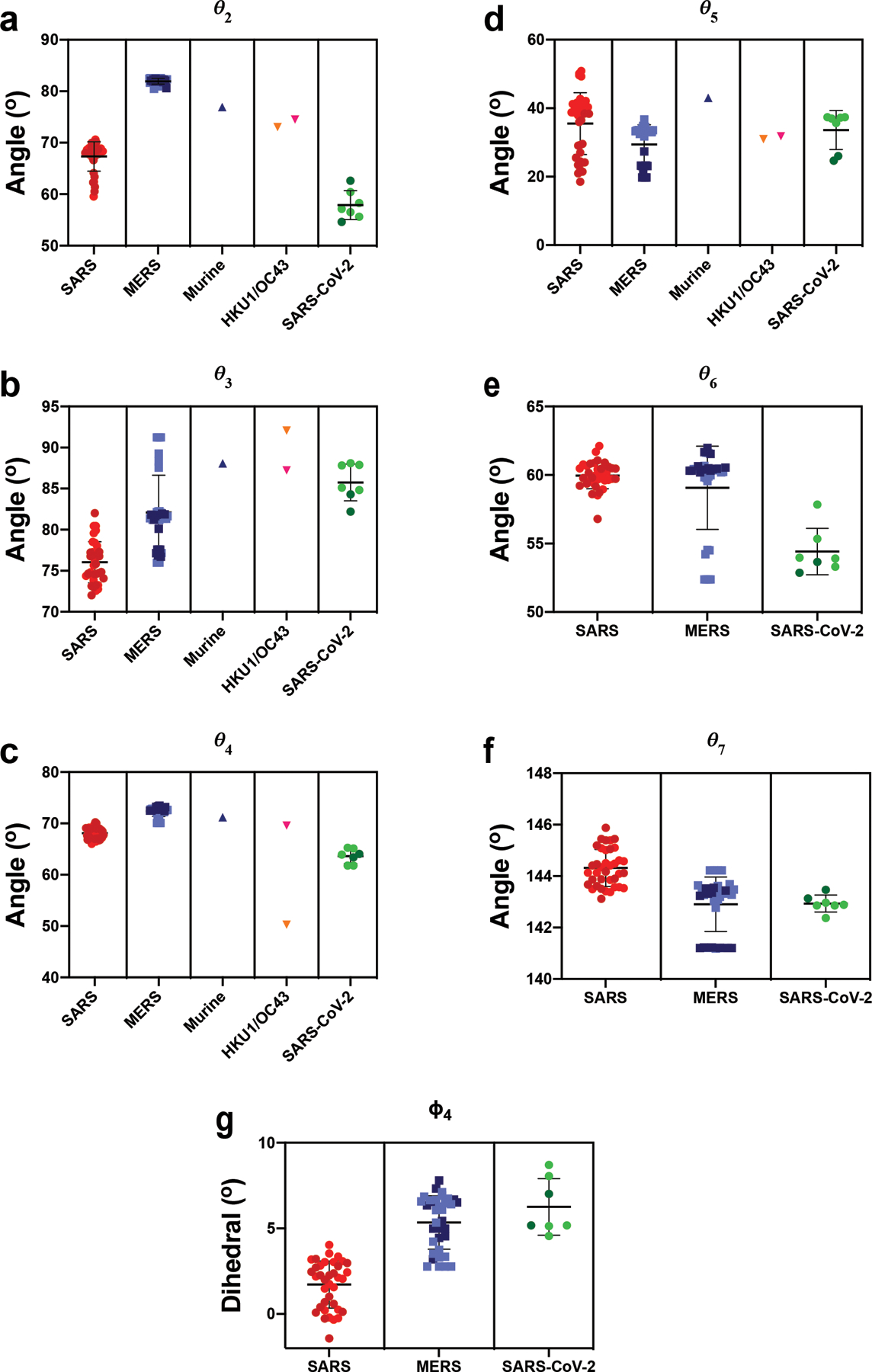
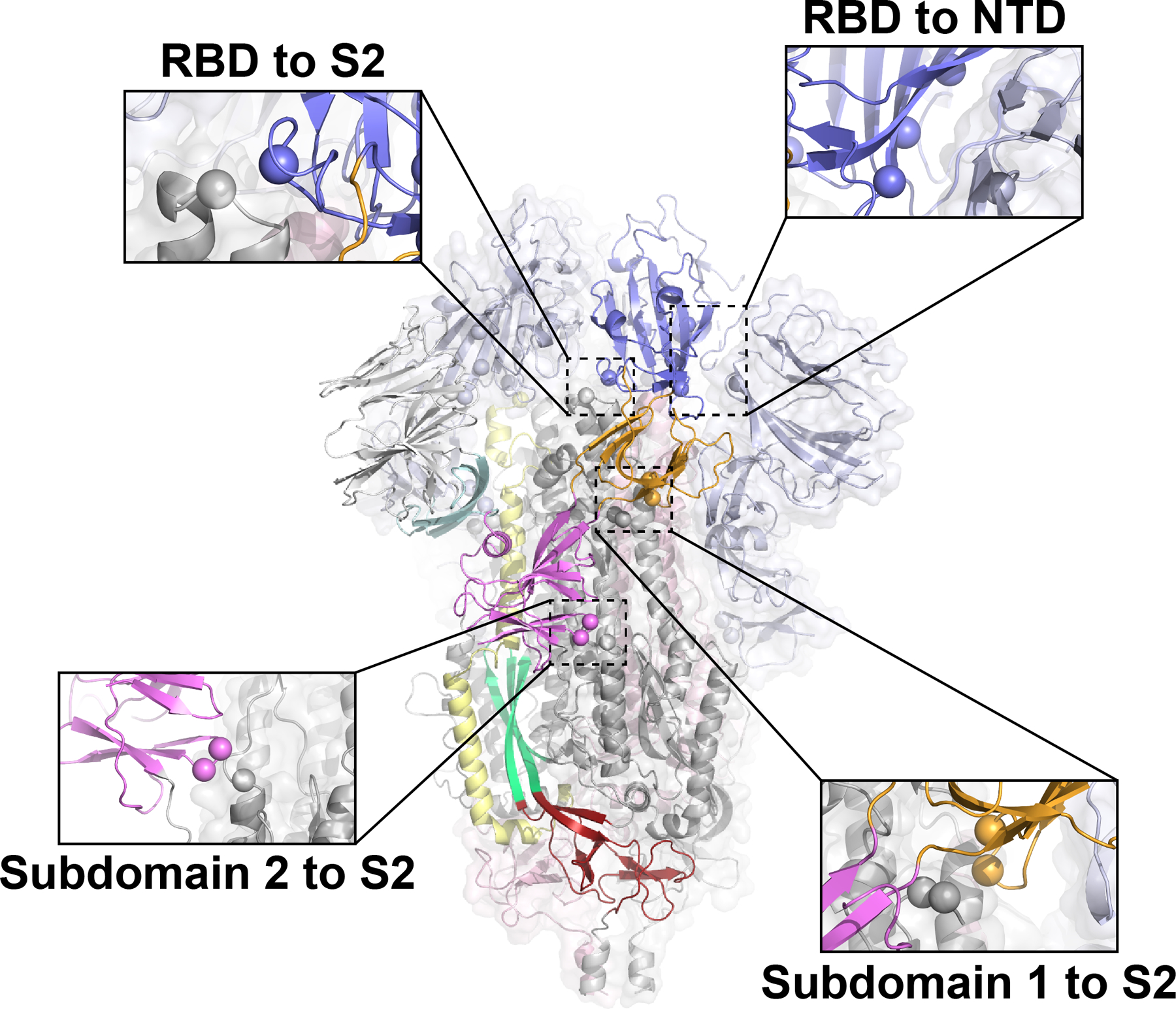
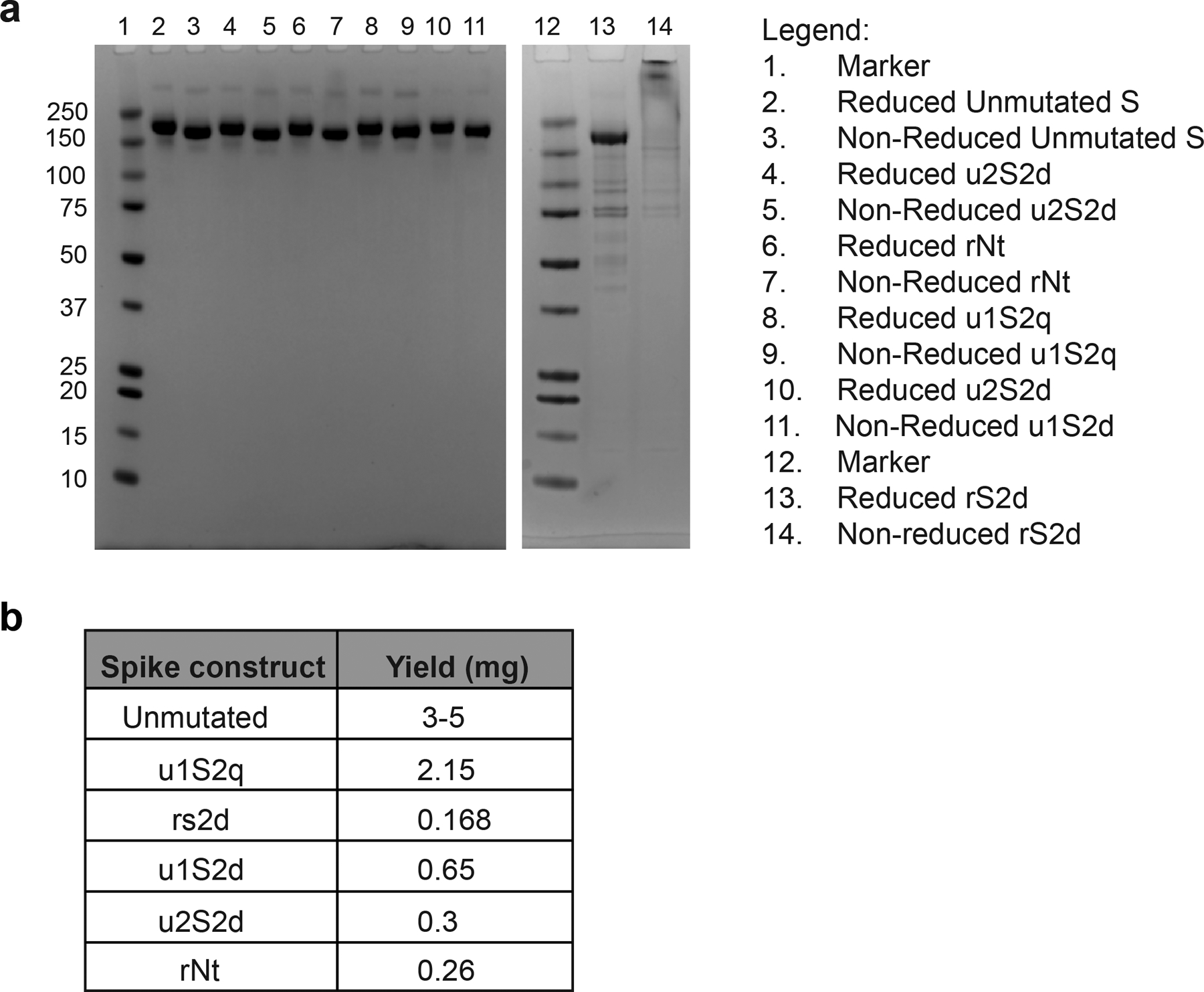
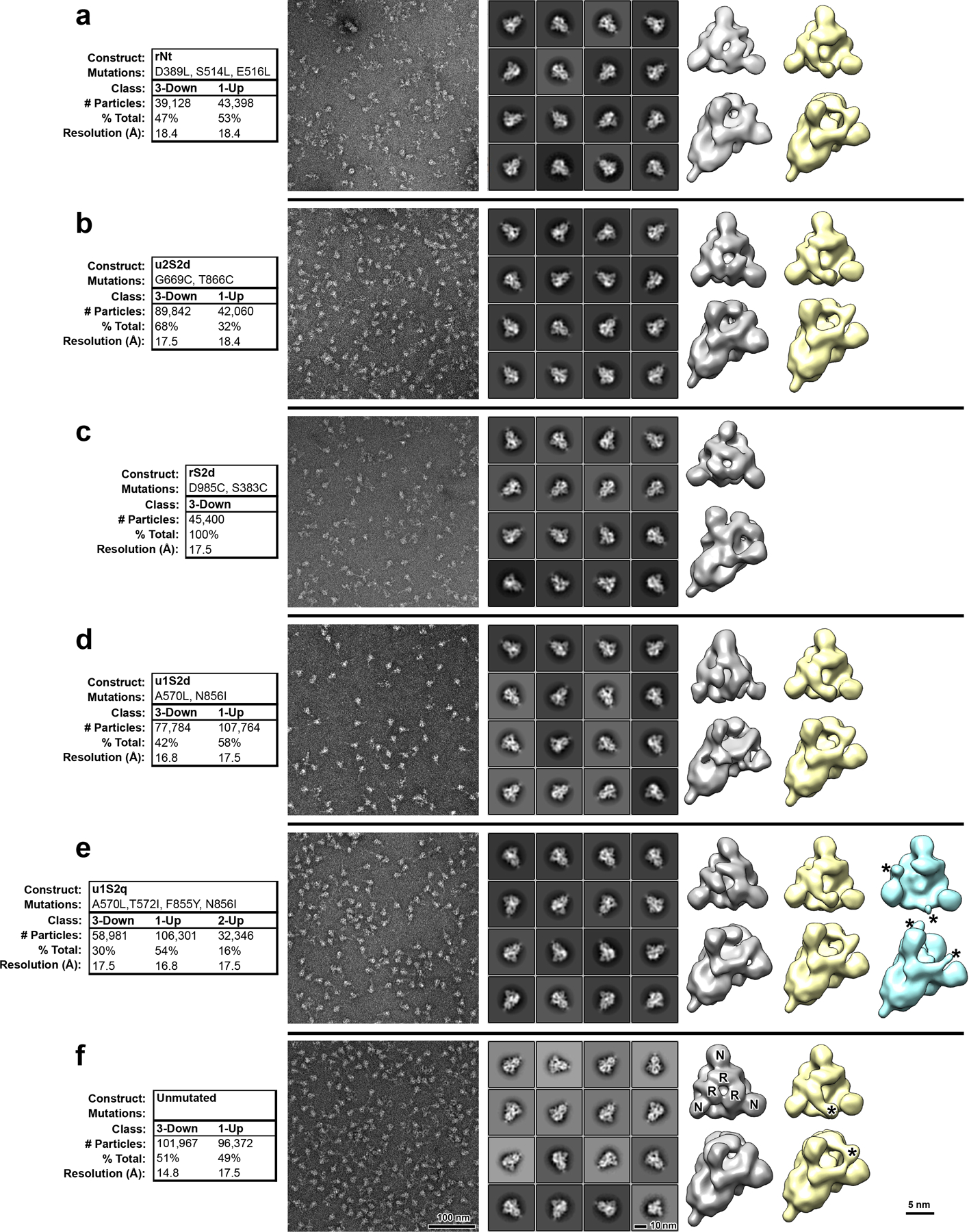
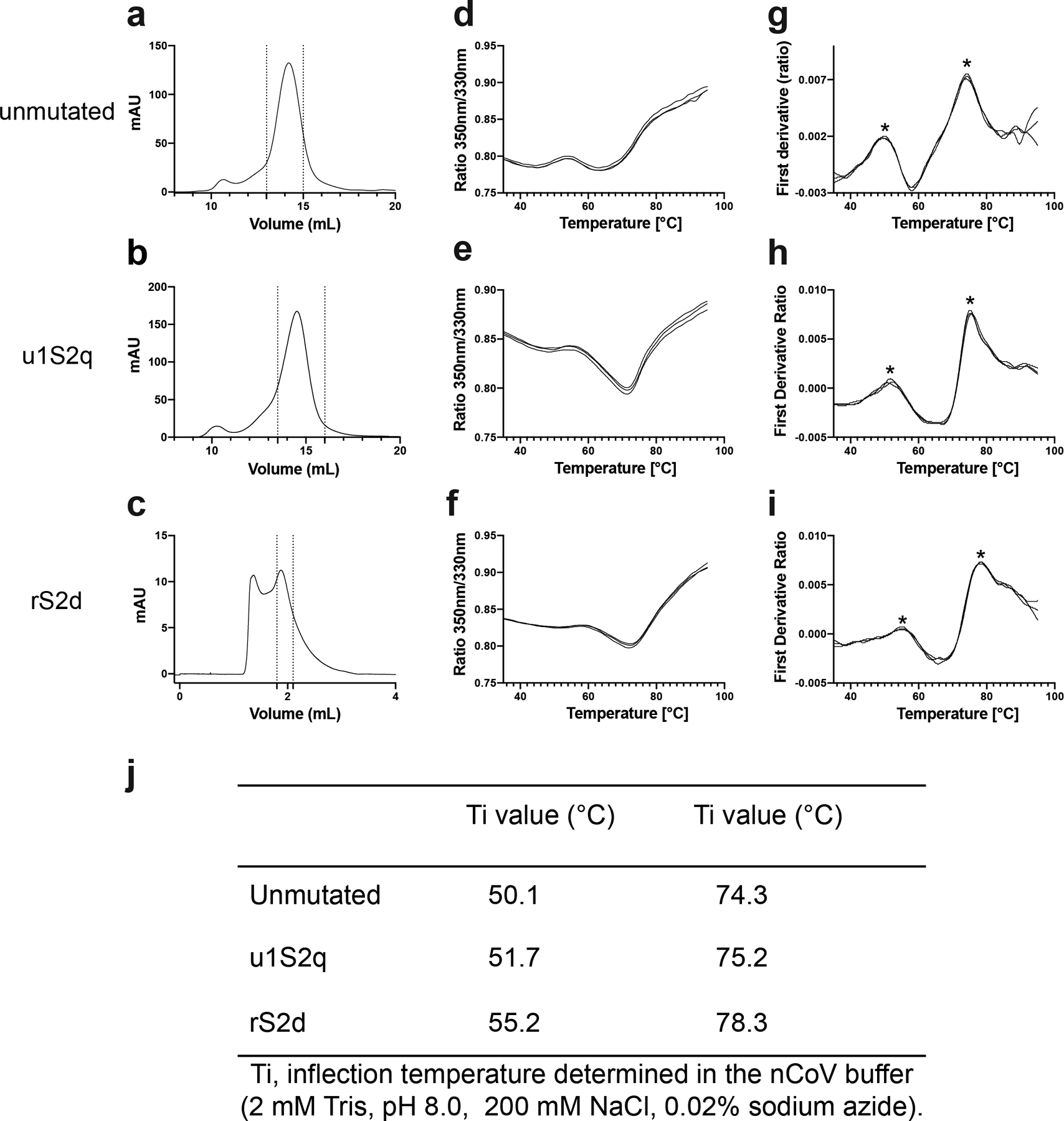
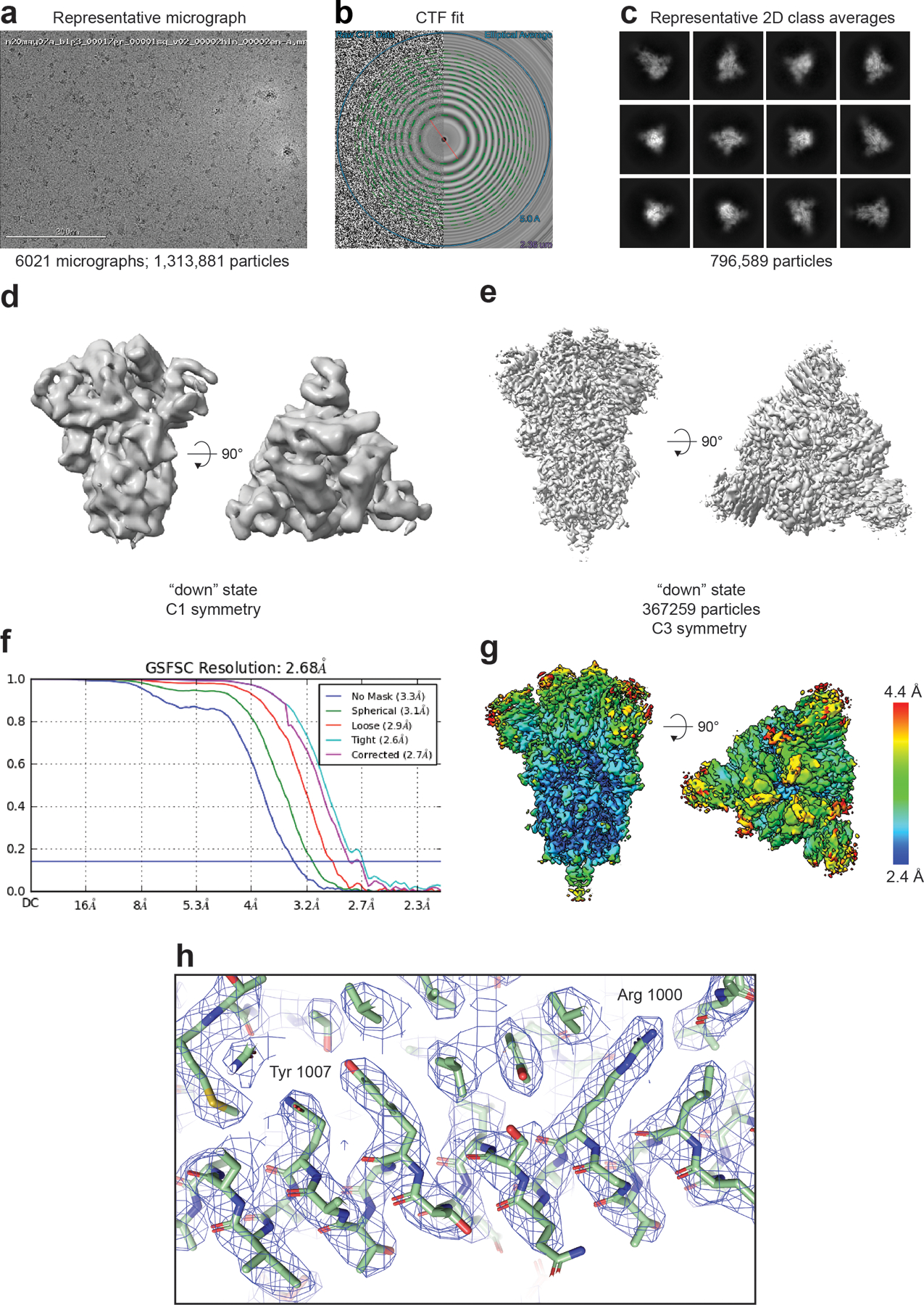
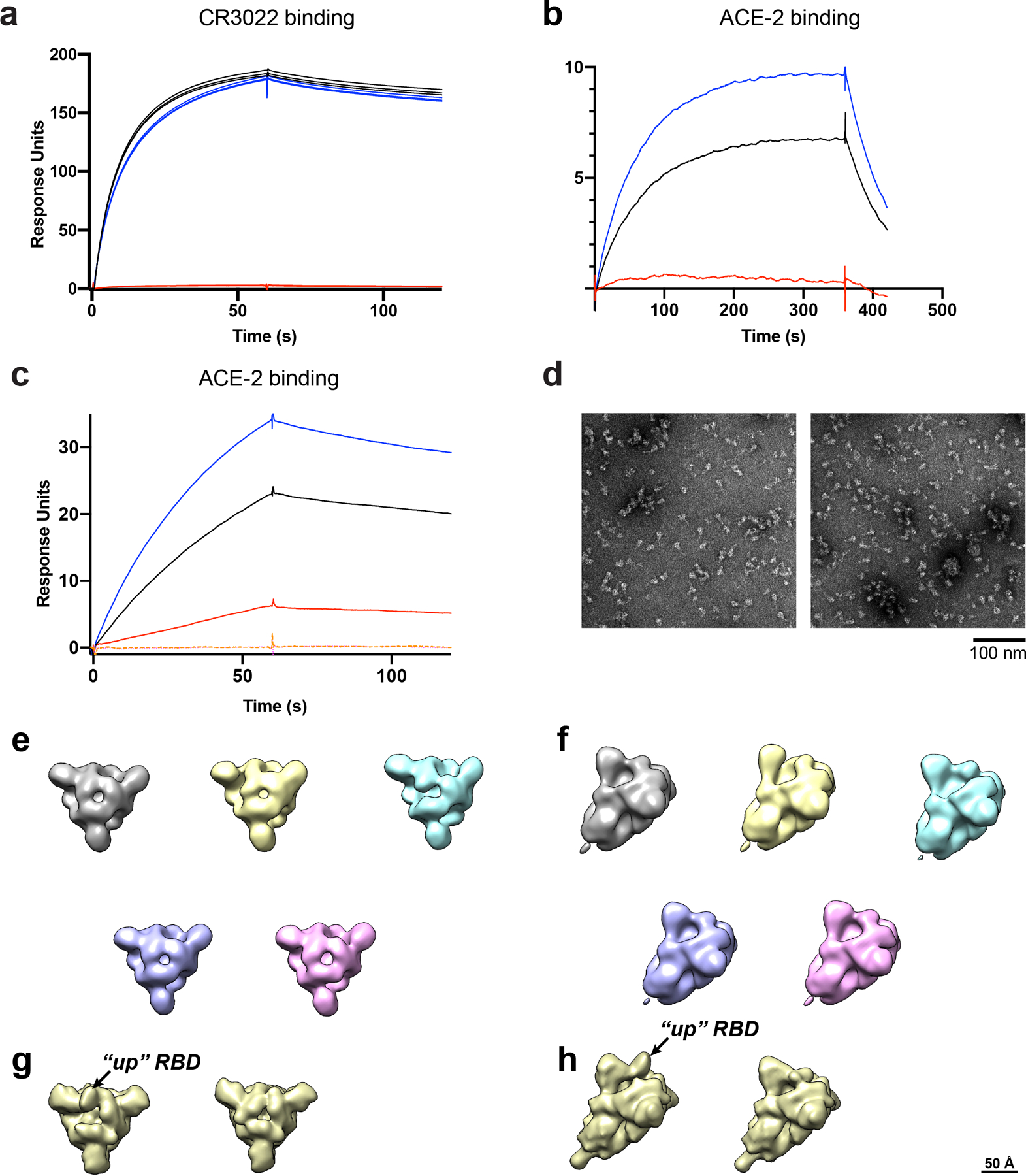
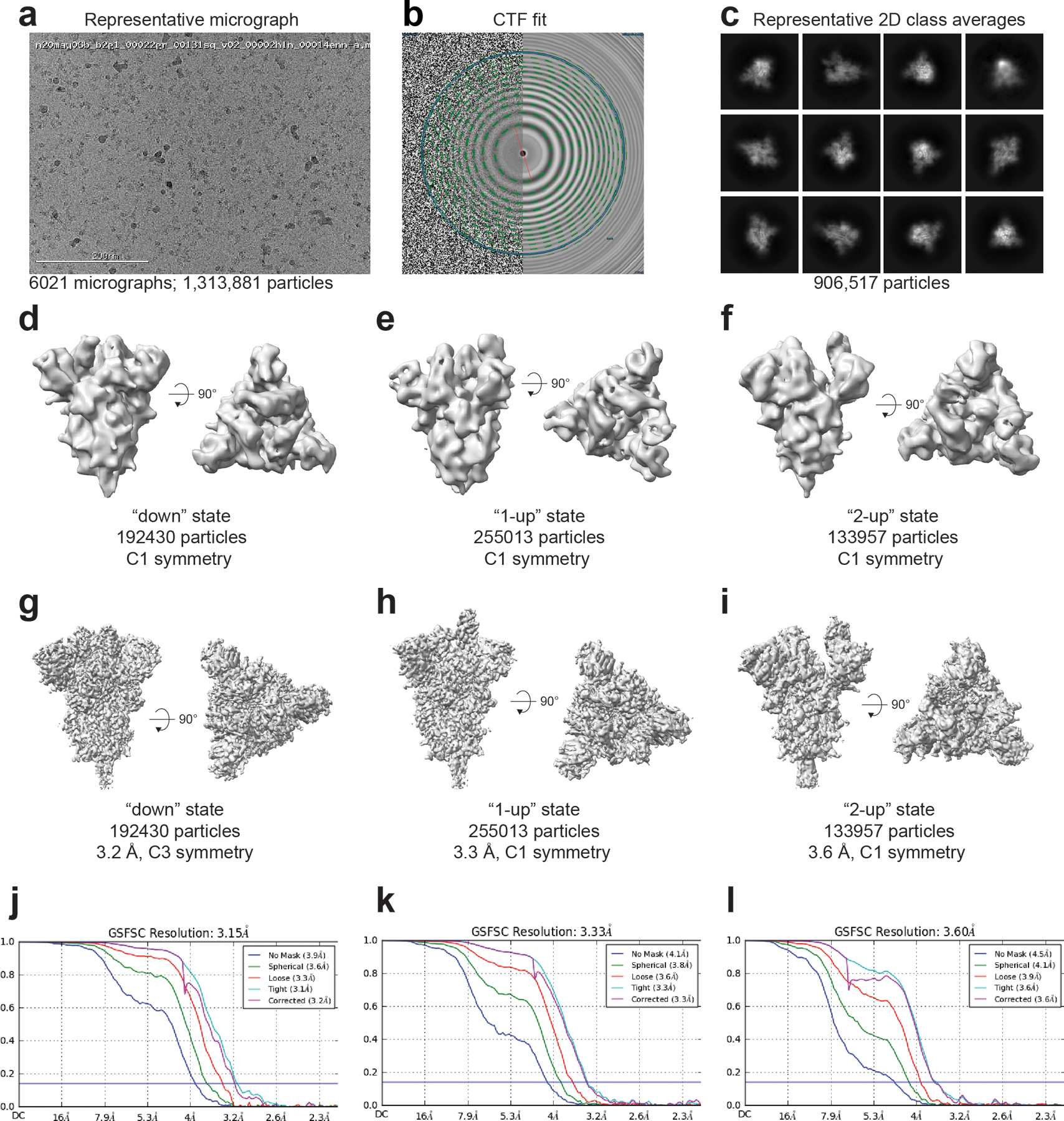
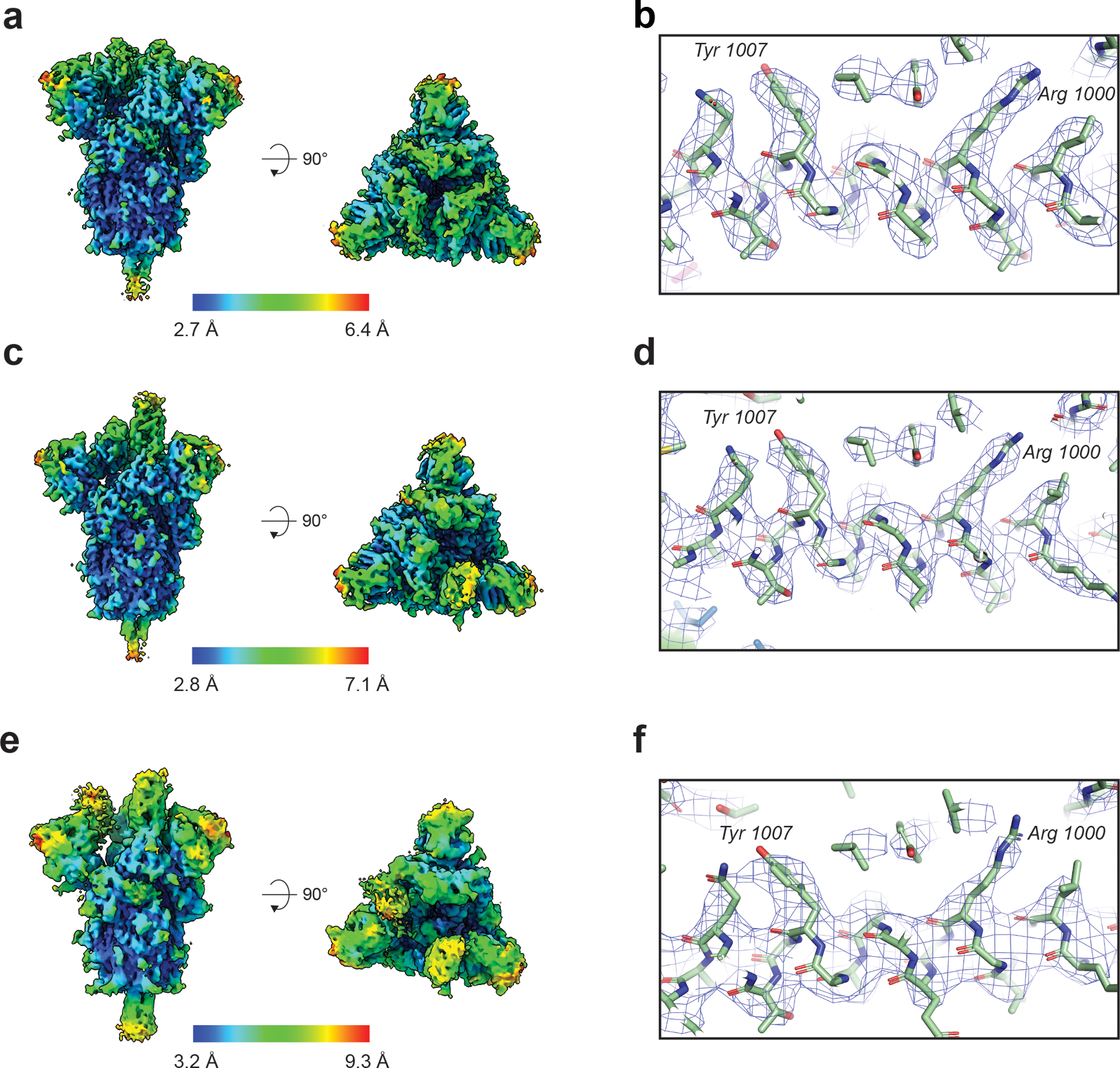
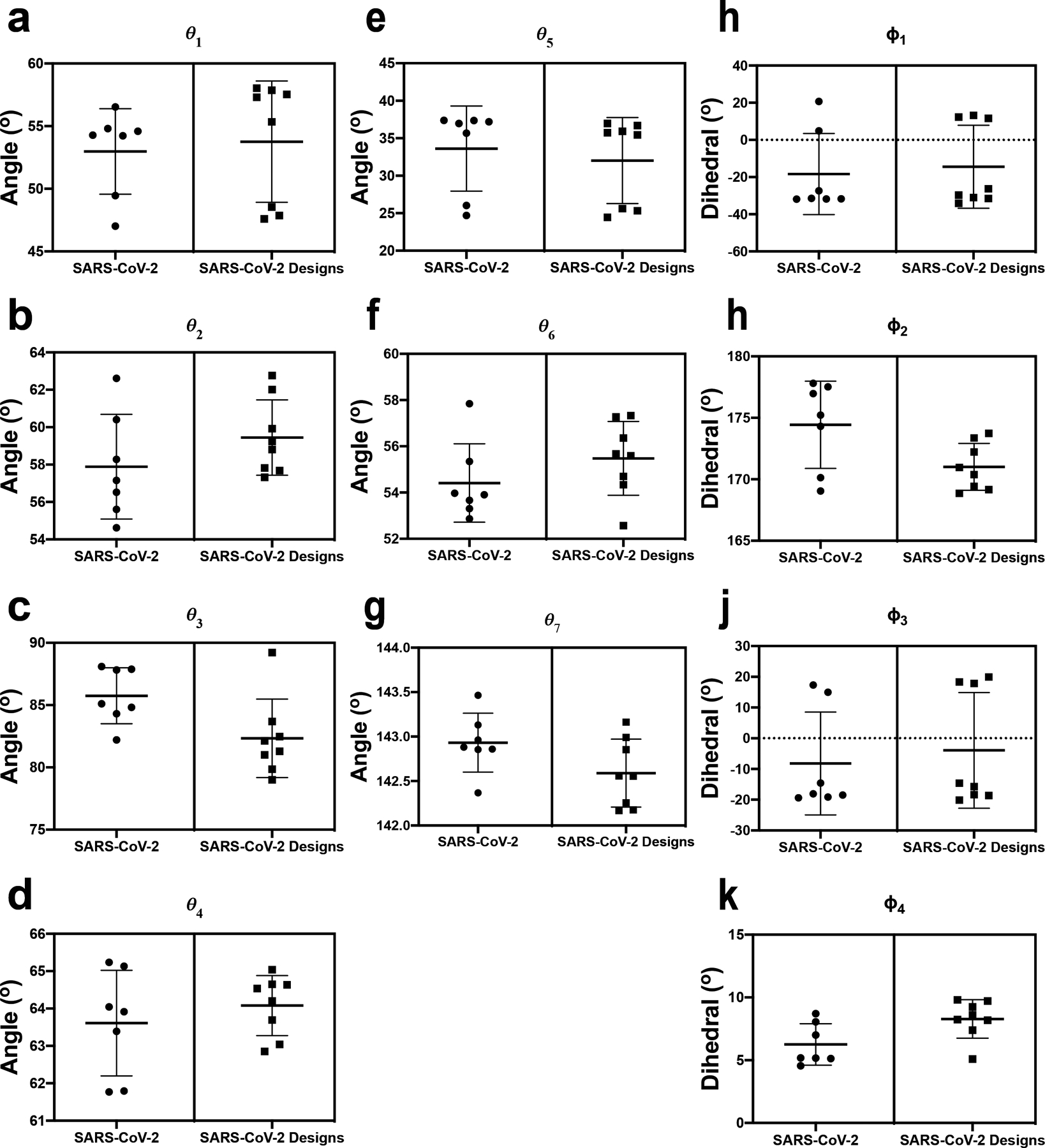
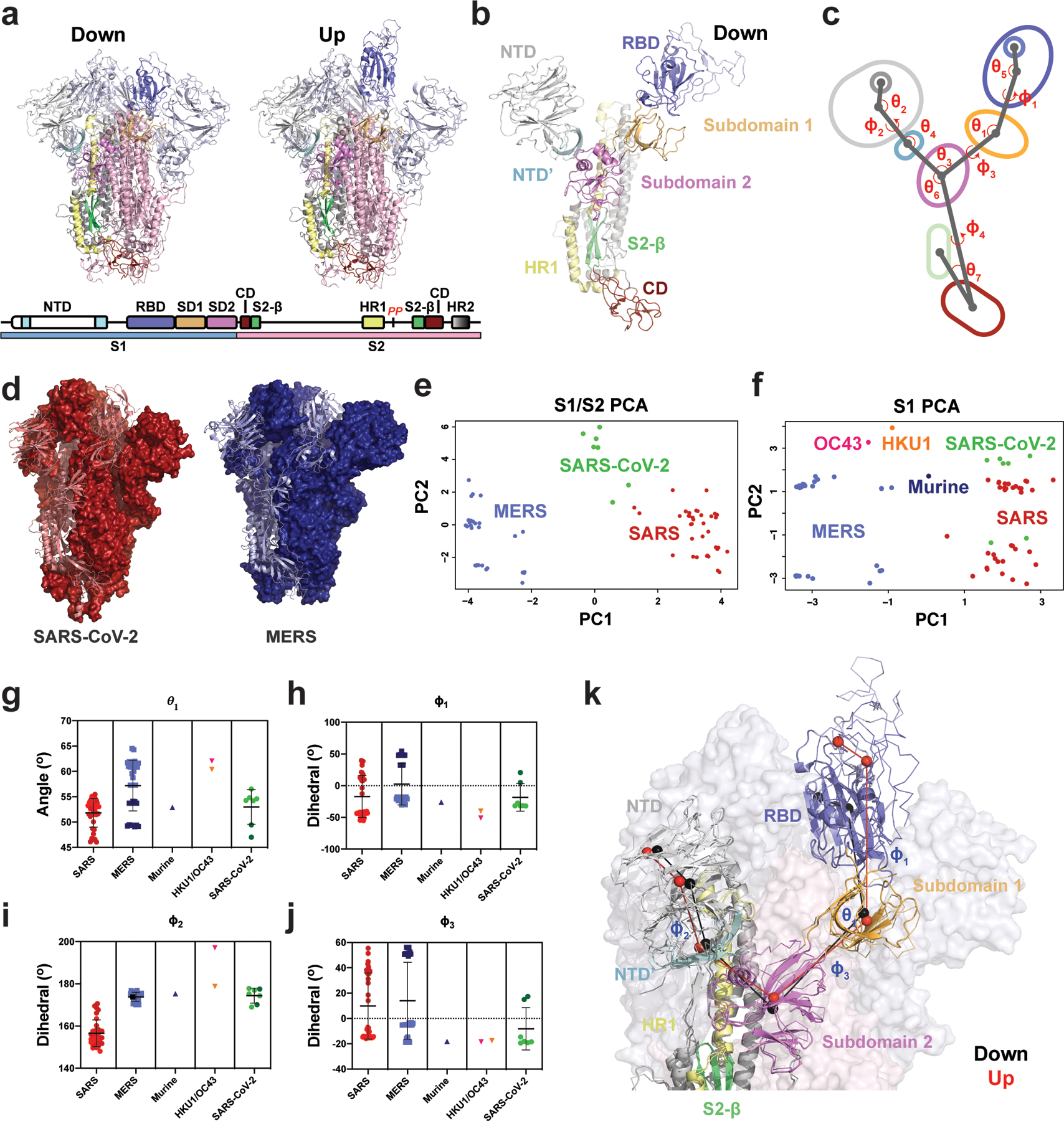
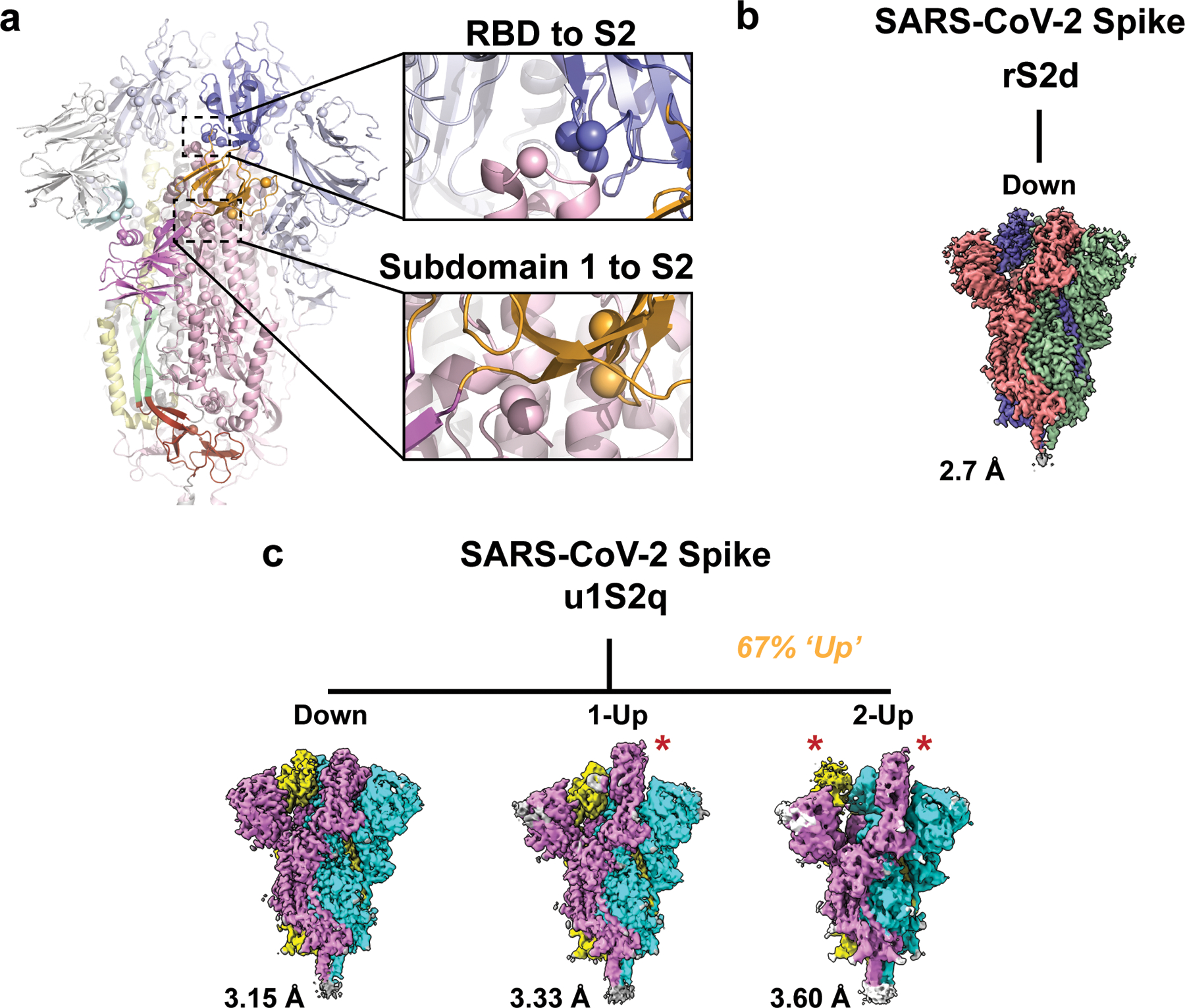
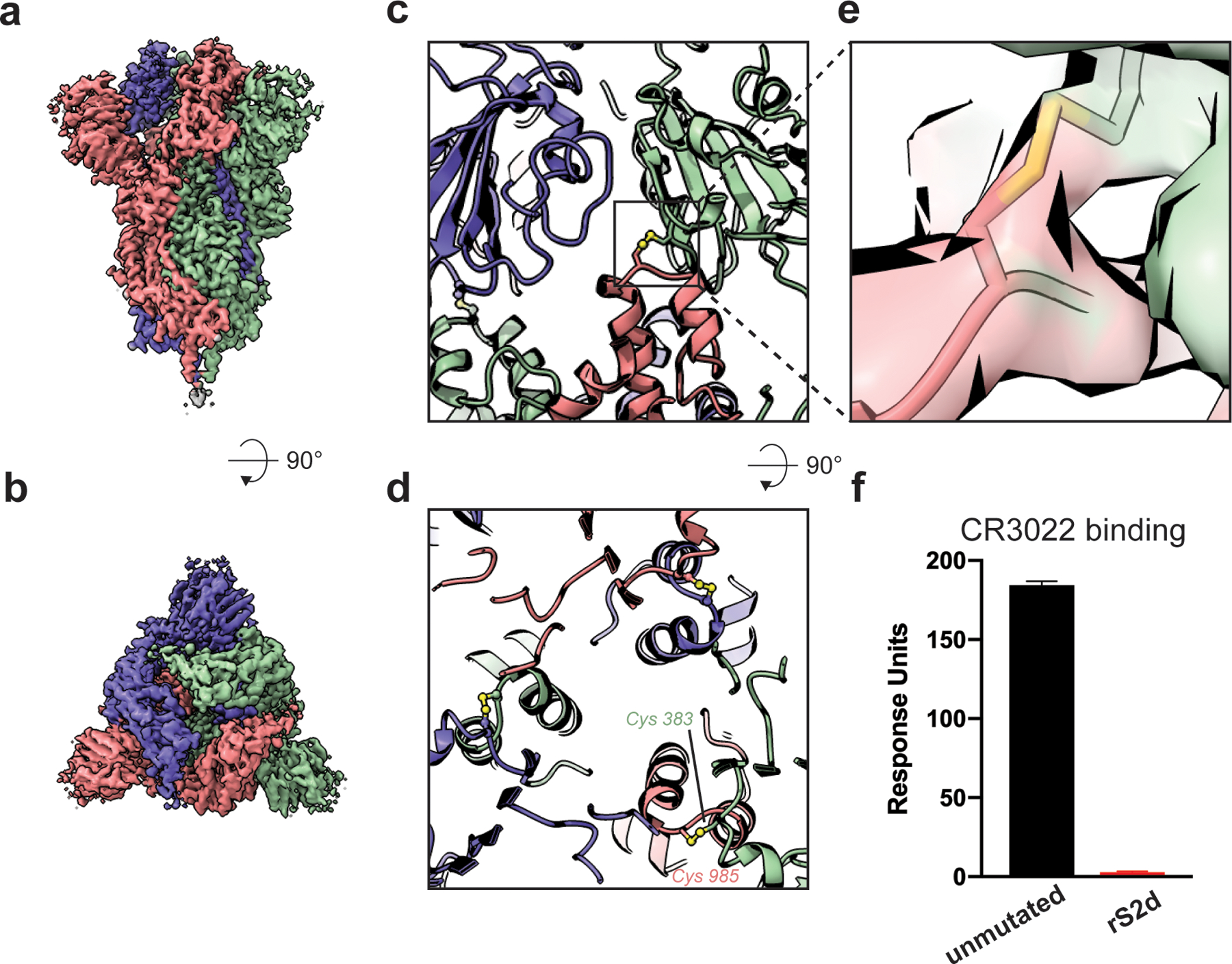
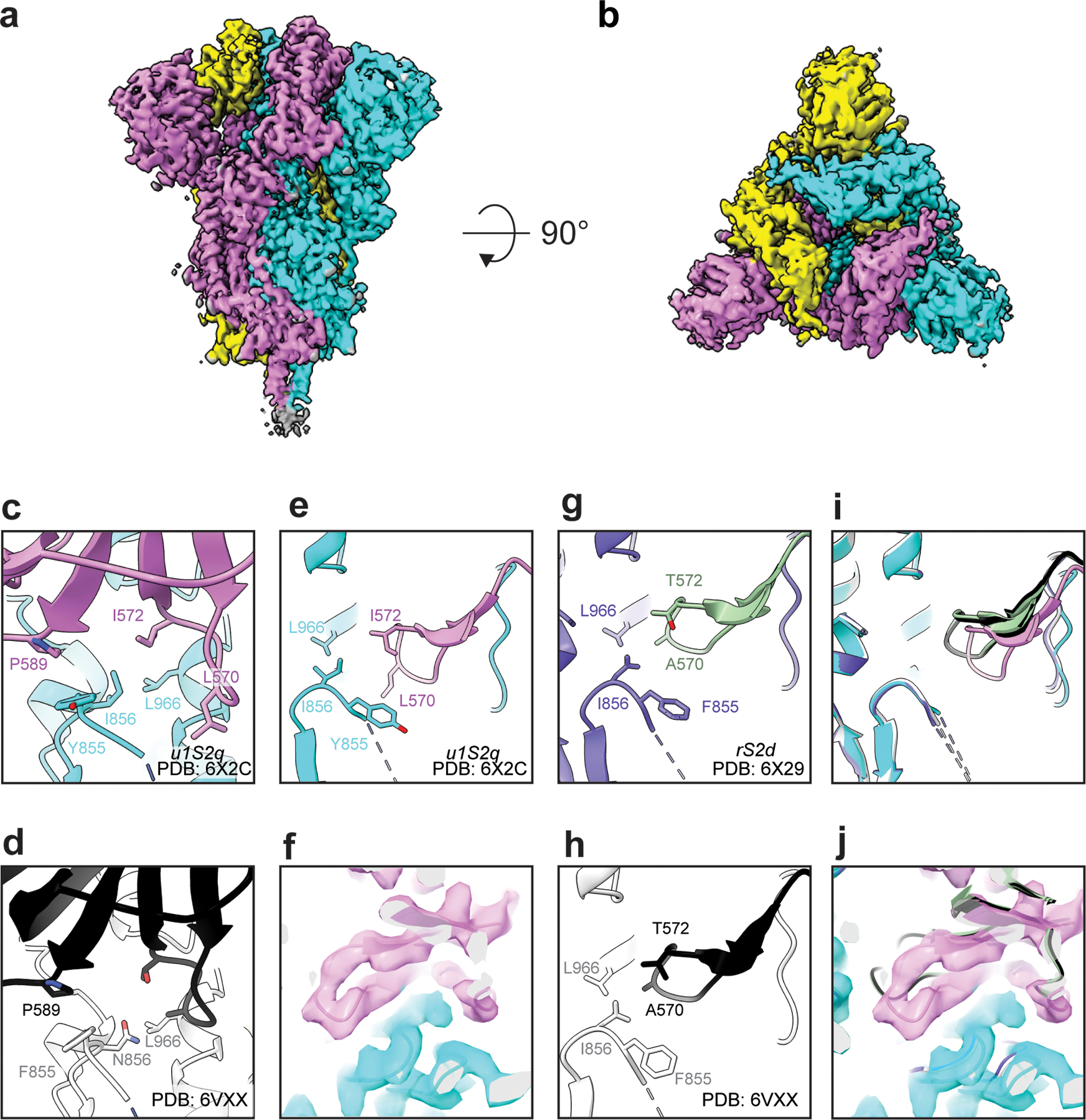
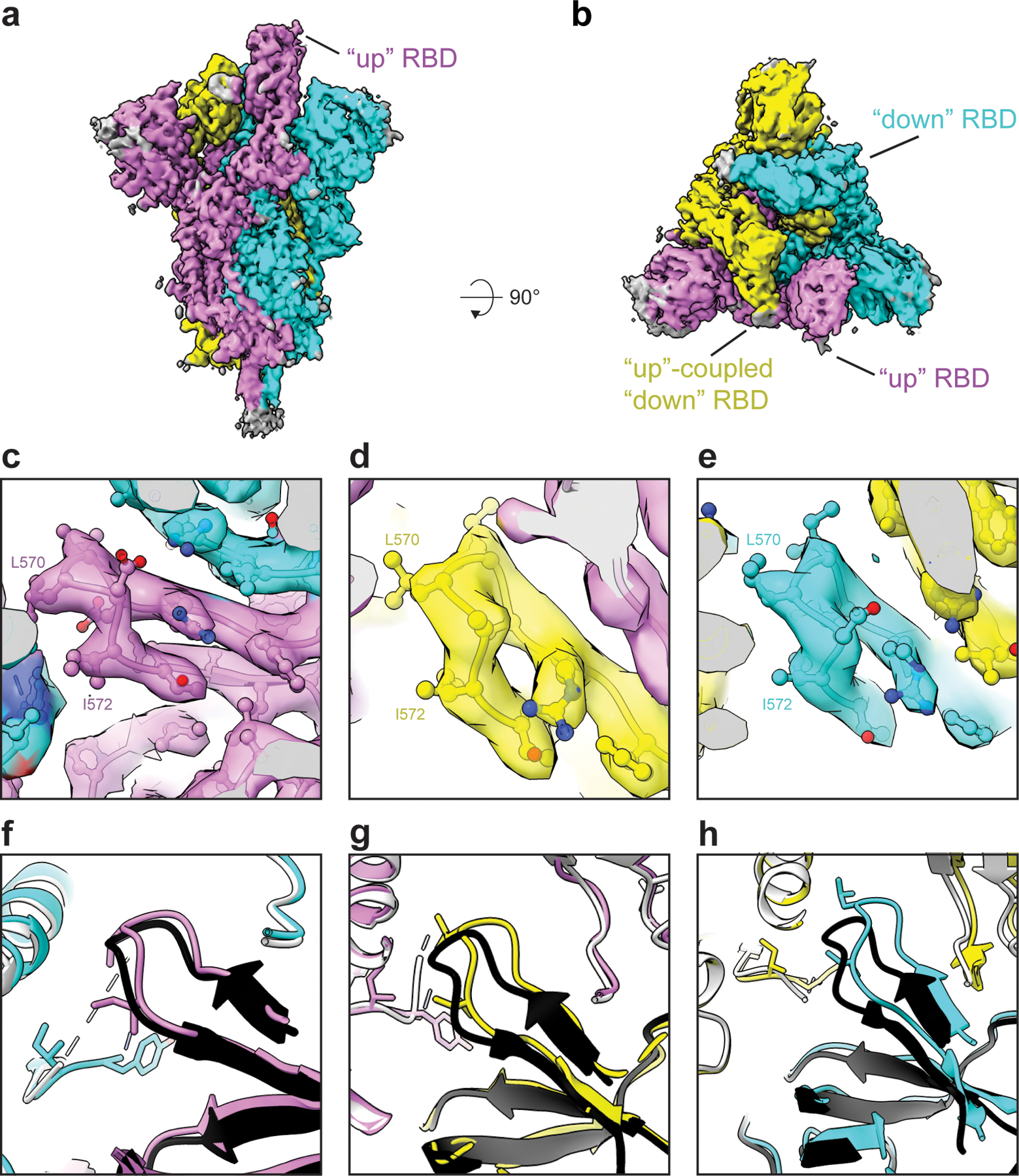
The coronavirus (CoV) spike (S) protein, involved in viral-host cell fusion, is the primary immunogenic target for virus neutralization and the current focus of many vaccine design efforts. The highly flexible S-protein, with its mobile domains, presents a moving target to the immune system. Here, to better understand S-protein mobility, we implemented a structure-based vector analysis of available β-CoV S-protein structures. Despite an overall similarity in domain organization, we found that S-proteins from different β-CoVs display distinct configurations. Based on this analysis, we developed two soluble ectodomain constructs for the SARS-CoV-2 S-protein, in which the highly immunogenic and mobile receptor binding domain (RBD) is either locked in the all-RBDs 'down' position or adopts 'up' state conformations more readily than the wild-type S-protein. These results demonstrate that the conformation of the S-protein can be controlled via rational design and can provide a framework for the development of engineered CoV S-proteins for vaccine applications.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Update of
-
Controlling the SARS-CoV-2 Spike Glycoprotein Conformation.bioRxiv [Preprint]. 2020 May 18:2020.05.18.102087. doi: 10.1101/2020.05.18.102087. bioRxiv. 2020. Update in: Nat Struct Mol Biol. 2020 Oct;27(10):925-933. doi: 10.1038/s41594-020-0479-4. PMID: 32511343 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
