

Signalling input from divergent pathways subverts B cell transformation
- PMID: 32699415
- PMCID: PMC7394729
- DOI: 10.1038/s41586-020-2513-4
Signalling input from divergent pathways subverts B cell transformation
Abstract
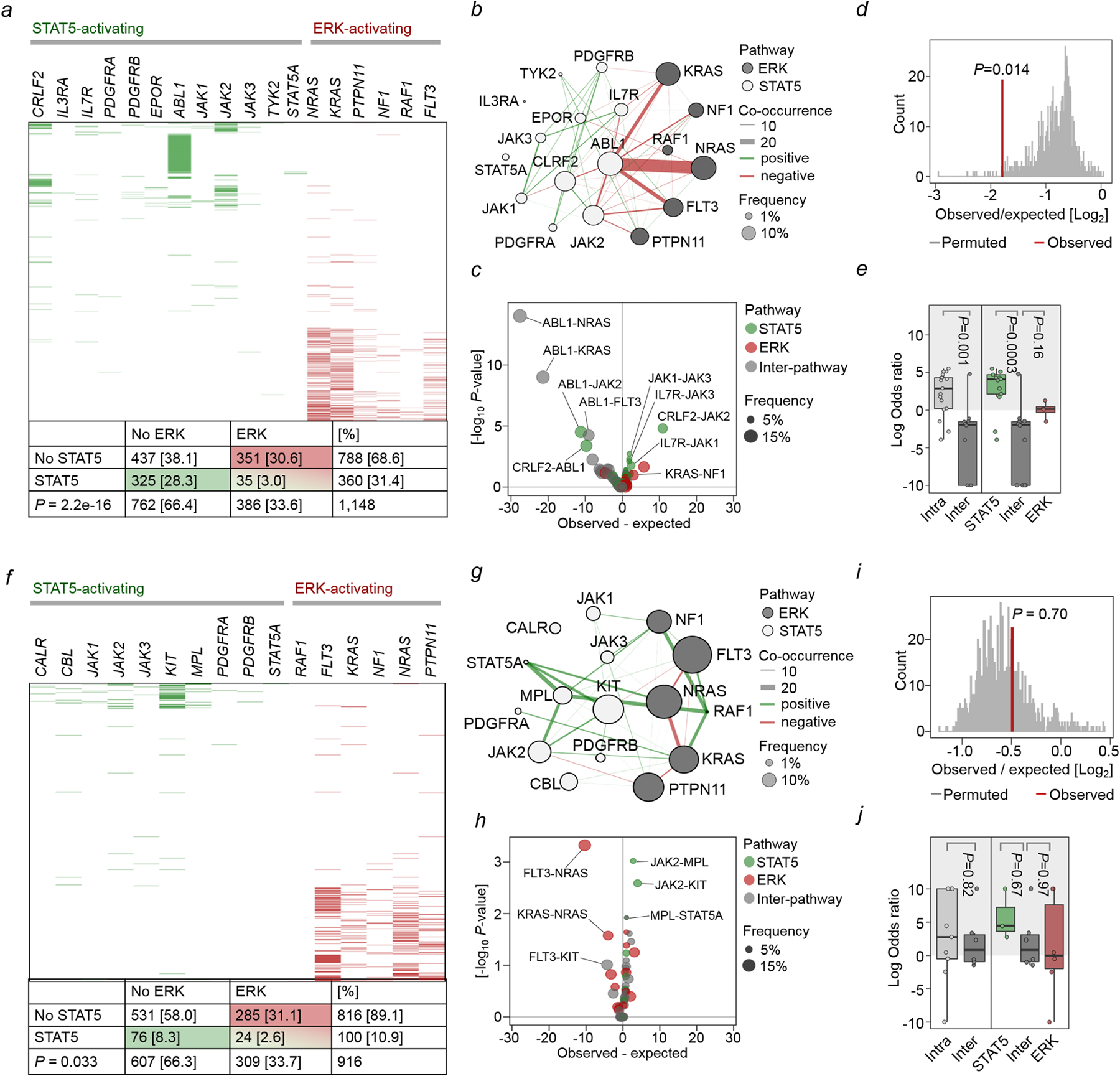
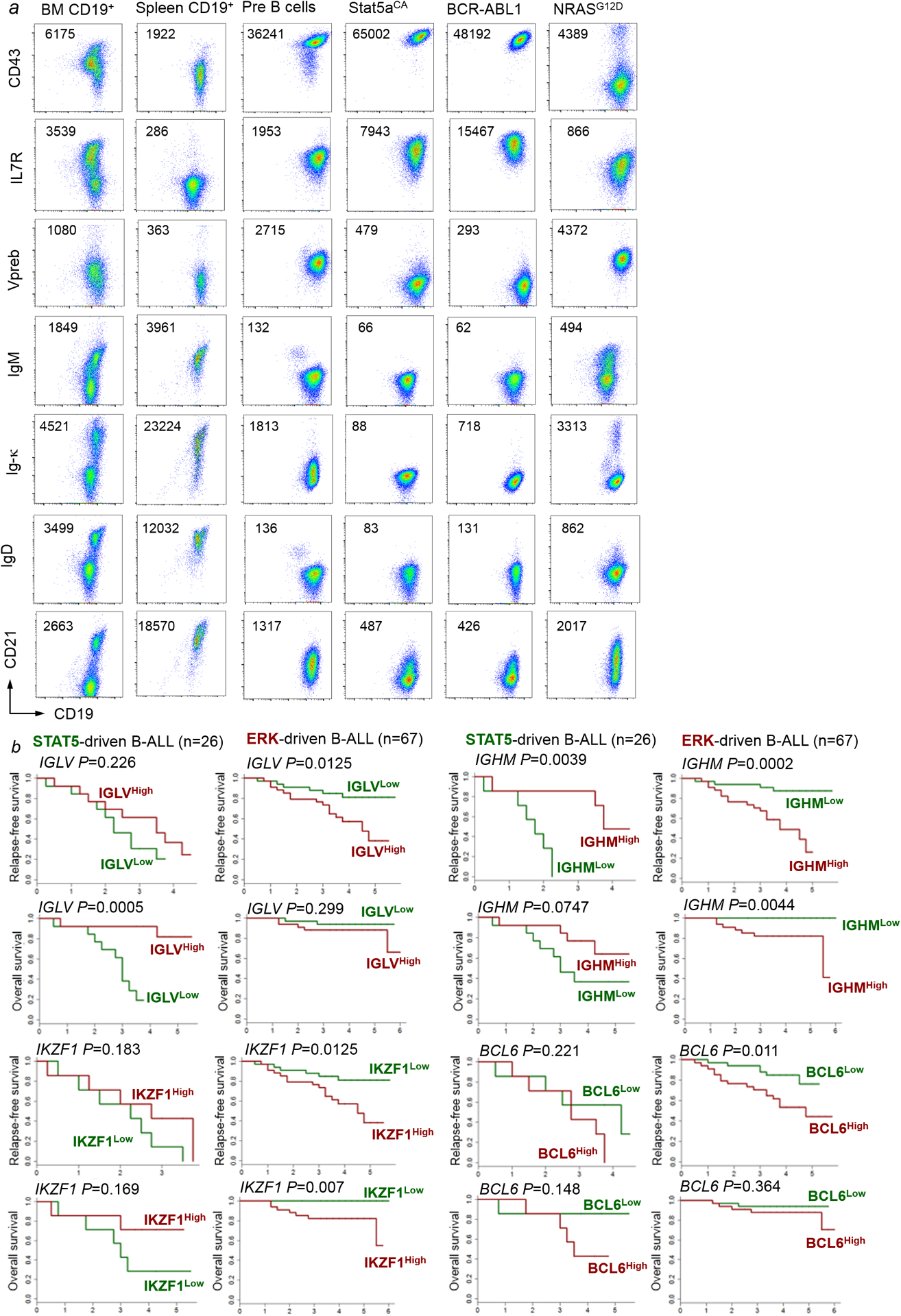
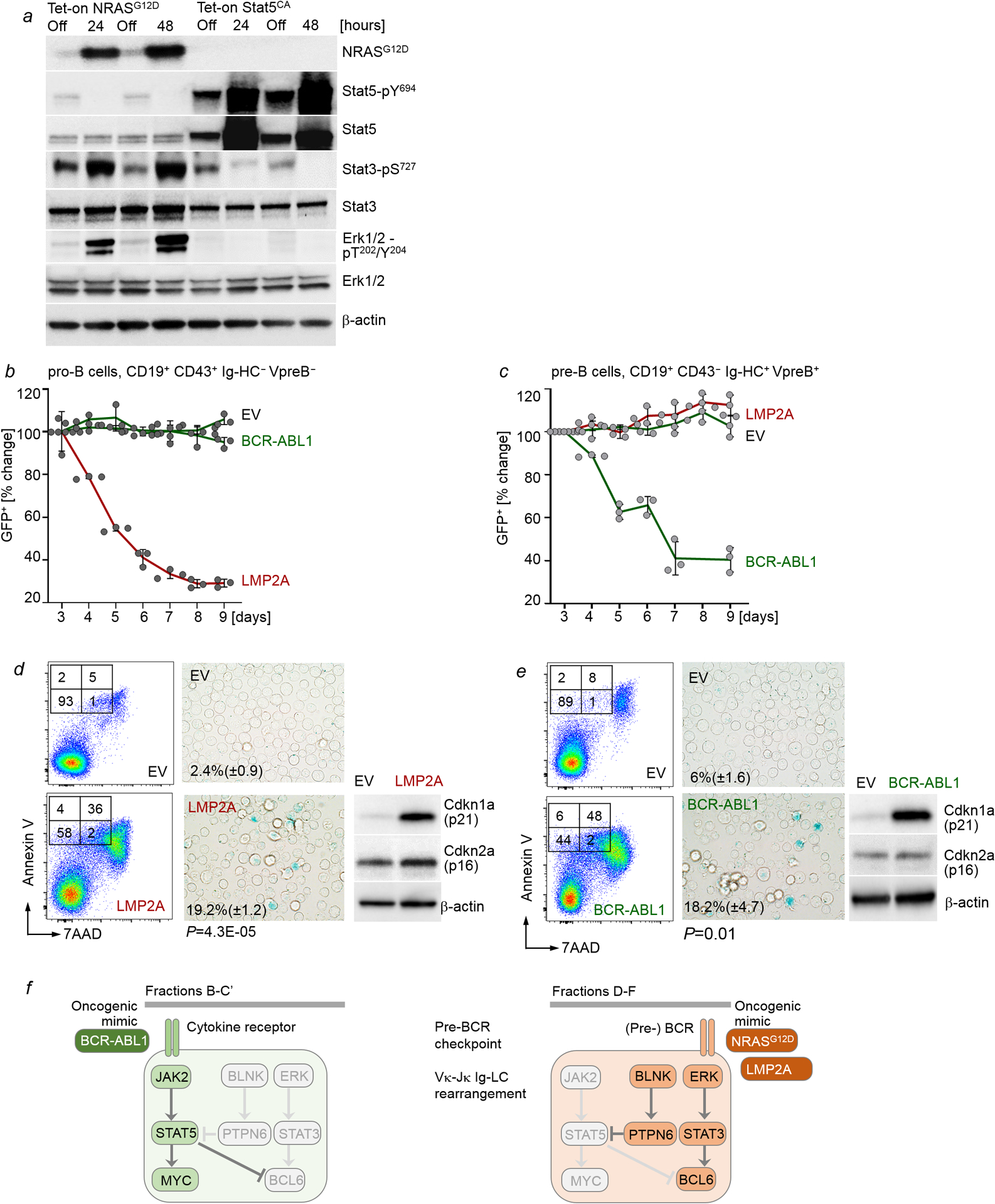
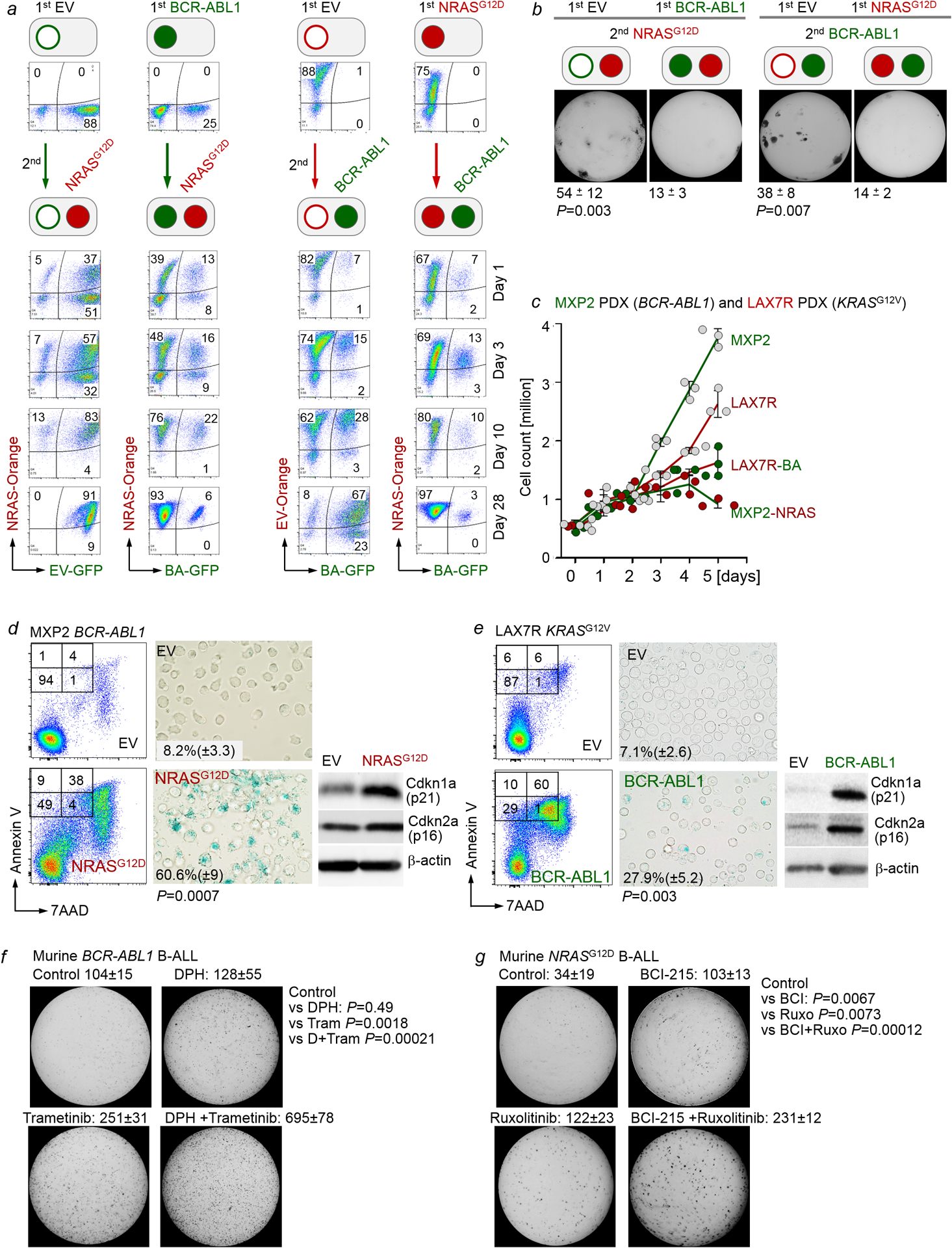
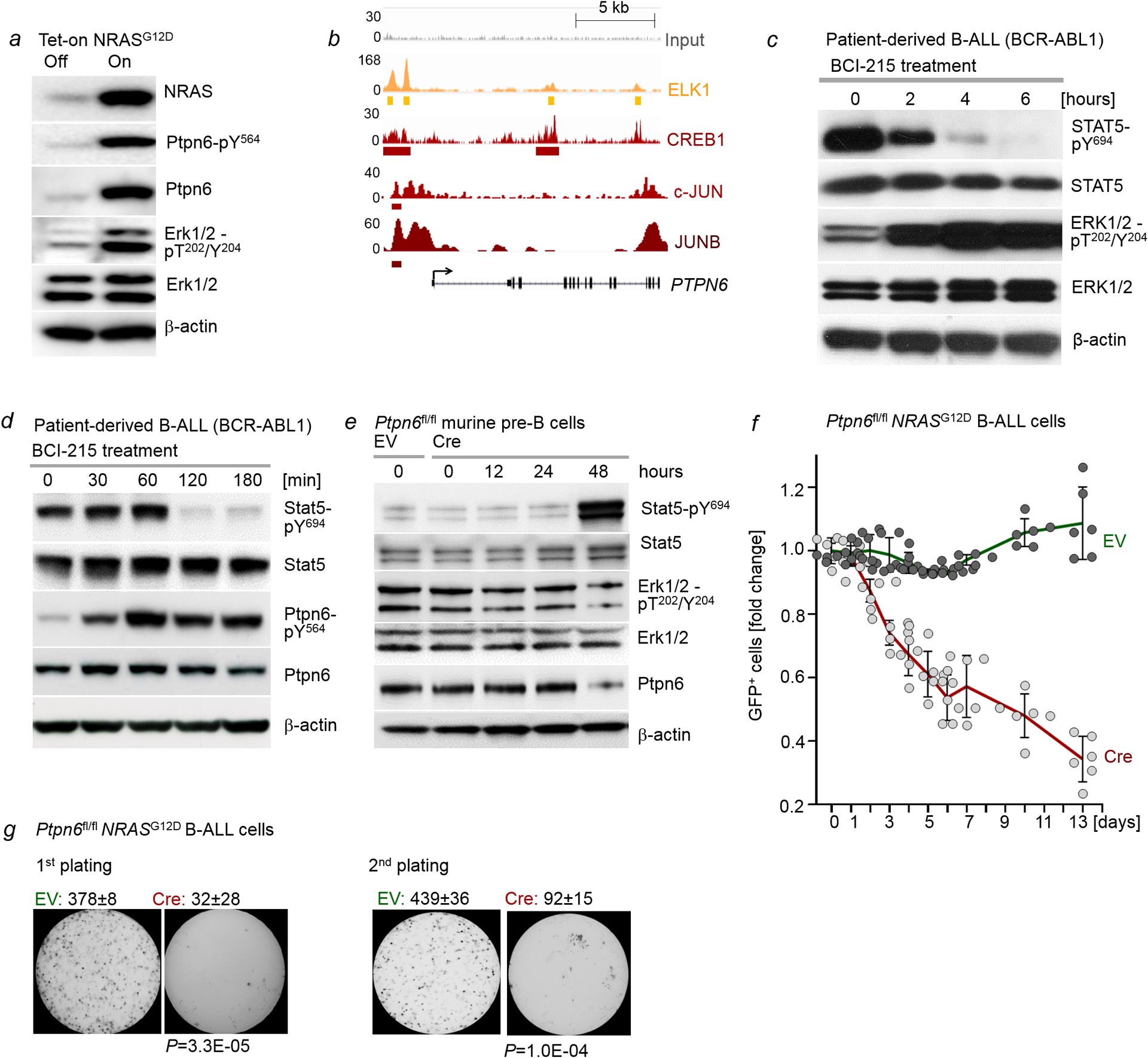
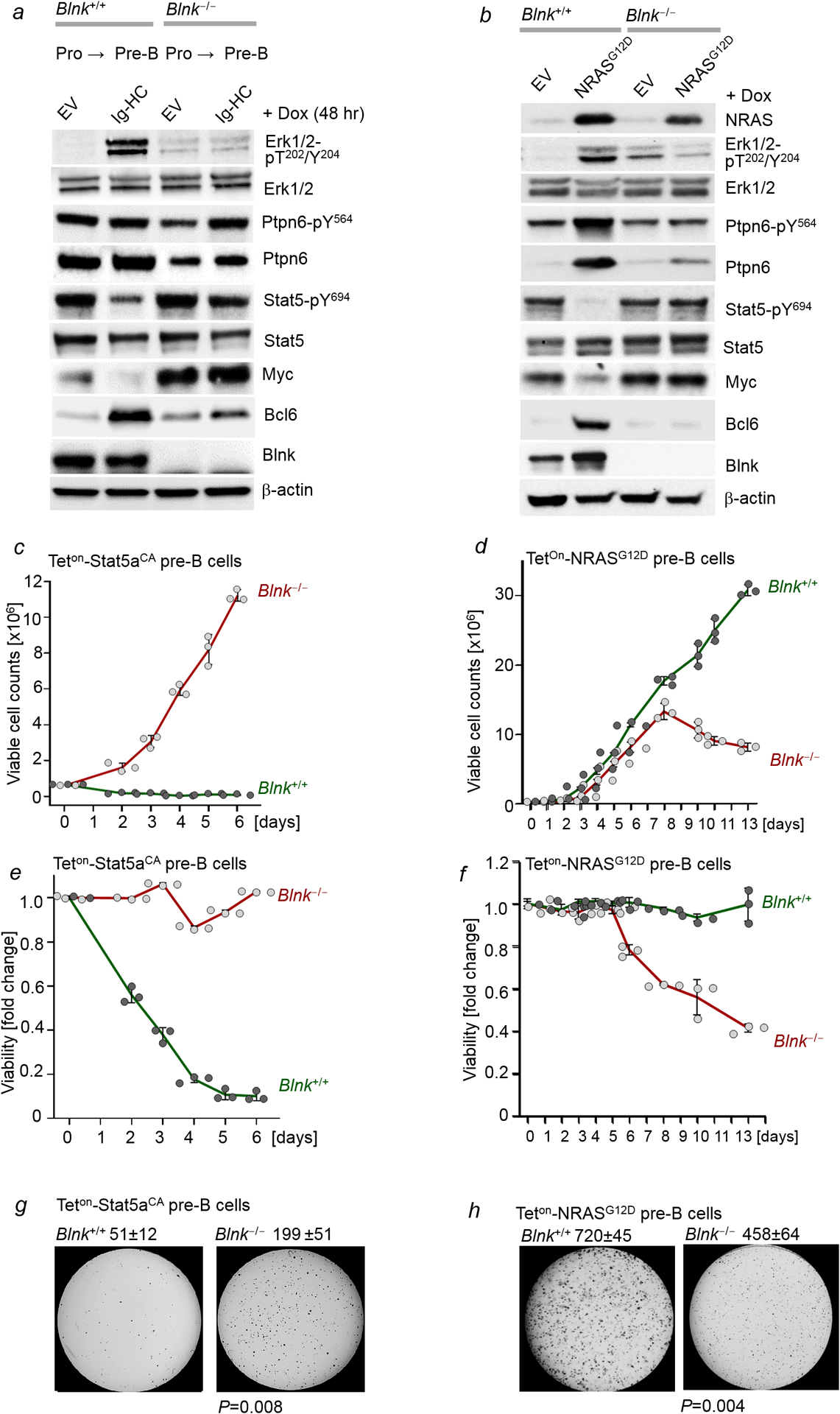
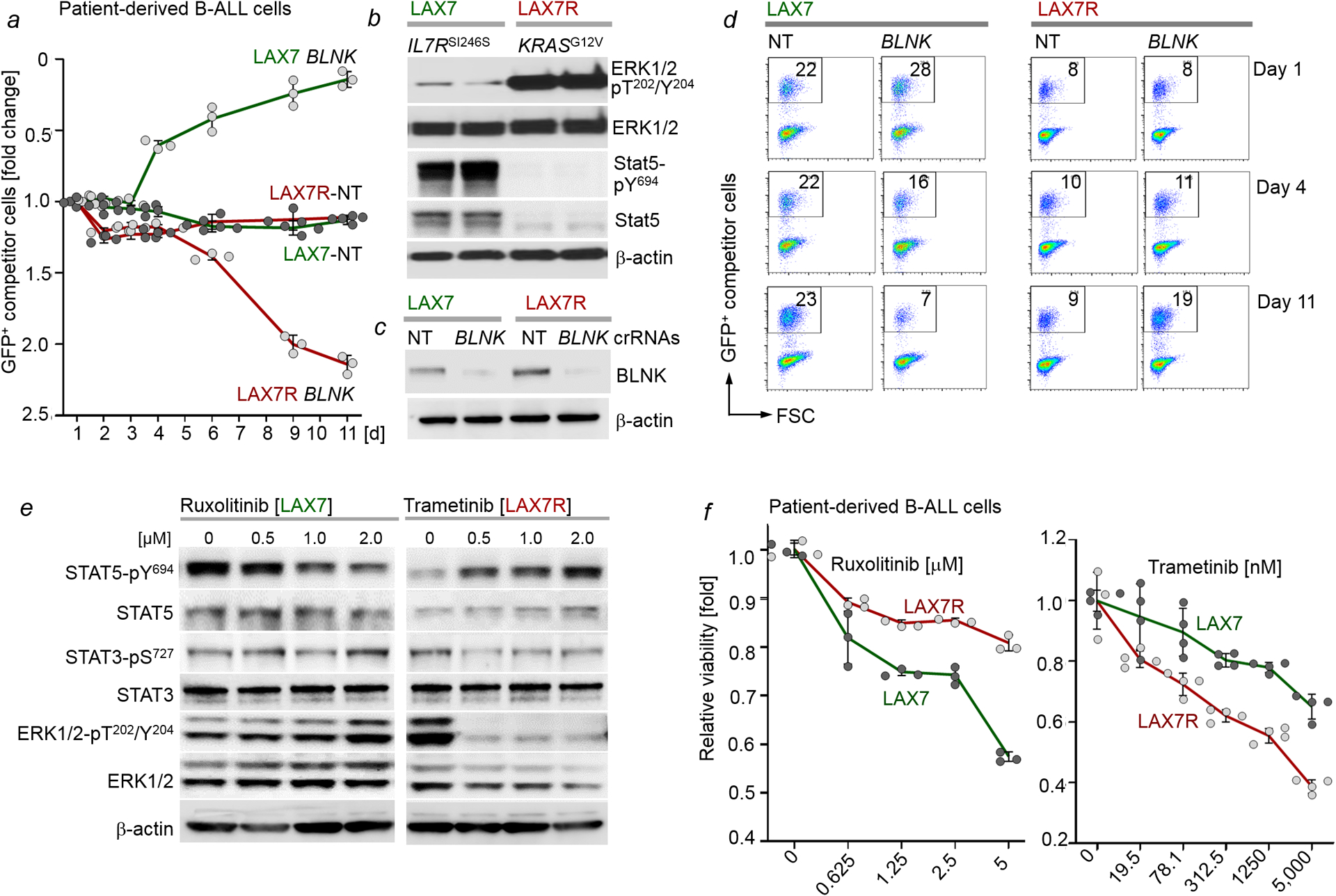
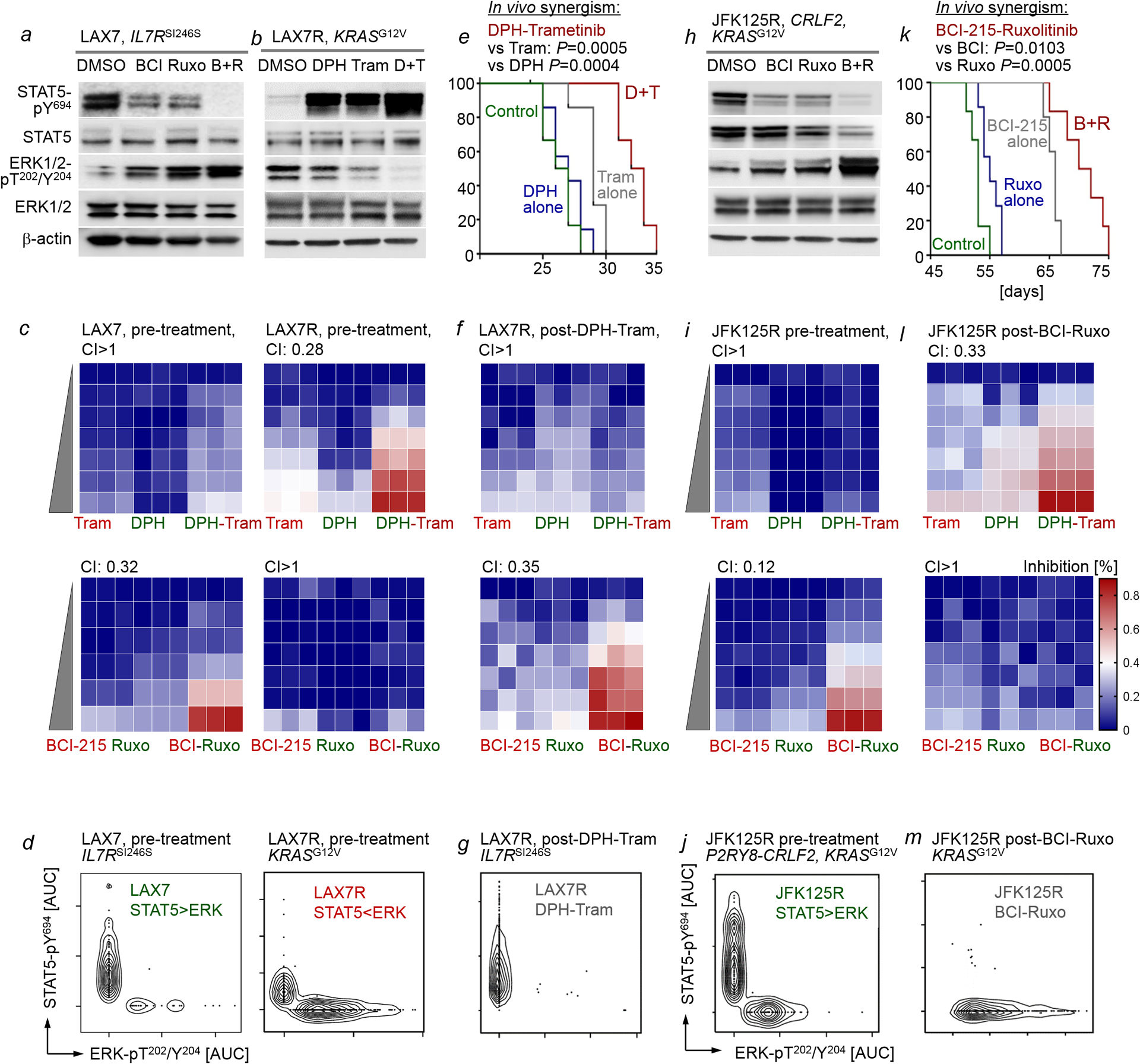
Malignant transformation of cells typically involves several genetic lesions, whose combined activity gives rise to cancer1. Here we analyse 1,148 patient-derived B-cell leukaemia (B-ALL) samples, and find that individual mutations do not promote leukaemogenesis unless they converge on one single oncogenic pathway that is characteristic of the differentiation stage of transformed B cells. Mutations that are not aligned with this central oncogenic driver activate divergent pathways and subvert transformation. Oncogenic lesions in B-ALL frequently mimic signalling through cytokine receptors at the pro-B-cell stage (via activation of the signal-transduction protein STAT5)2-4 or pre-B-cell receptors in more mature cells (via activation of the protein kinase ERK)5-8. STAT5- and ERK-activating lesions are found frequently, but occur together in only around 3% of cases (P = 2.2 × 10-16). Single-cell mutation and phospho-protein analyses reveal the segregation of oncogenic STAT5 and ERK activation to competing clones. STAT5 and ERK engage opposing biochemical and transcriptional programs that are orchestrated by the transcription factors MYC and BCL6, respectively. Genetic reactivation of the divergent (suppressed) pathway comes at the expense of the principal oncogenic driver and reverses transformation. Conversely, deletion of divergent pathway components accelerates leukaemogenesis. Thus, persistence of divergent signalling pathways represents a powerful barrier to transformation, while convergence on one principal driver defines a central event in leukaemia initiation. Pharmacological reactivation of suppressed divergent circuits synergizes strongly with inhibition of the principal oncogenic driver. Hence, reactivation of divergent pathways can be leveraged as a previously unrecognized strategy to enhance treatment responses.
Conflict of interest statement
Declaration of Interests
The authors have no competing interests.
Figures


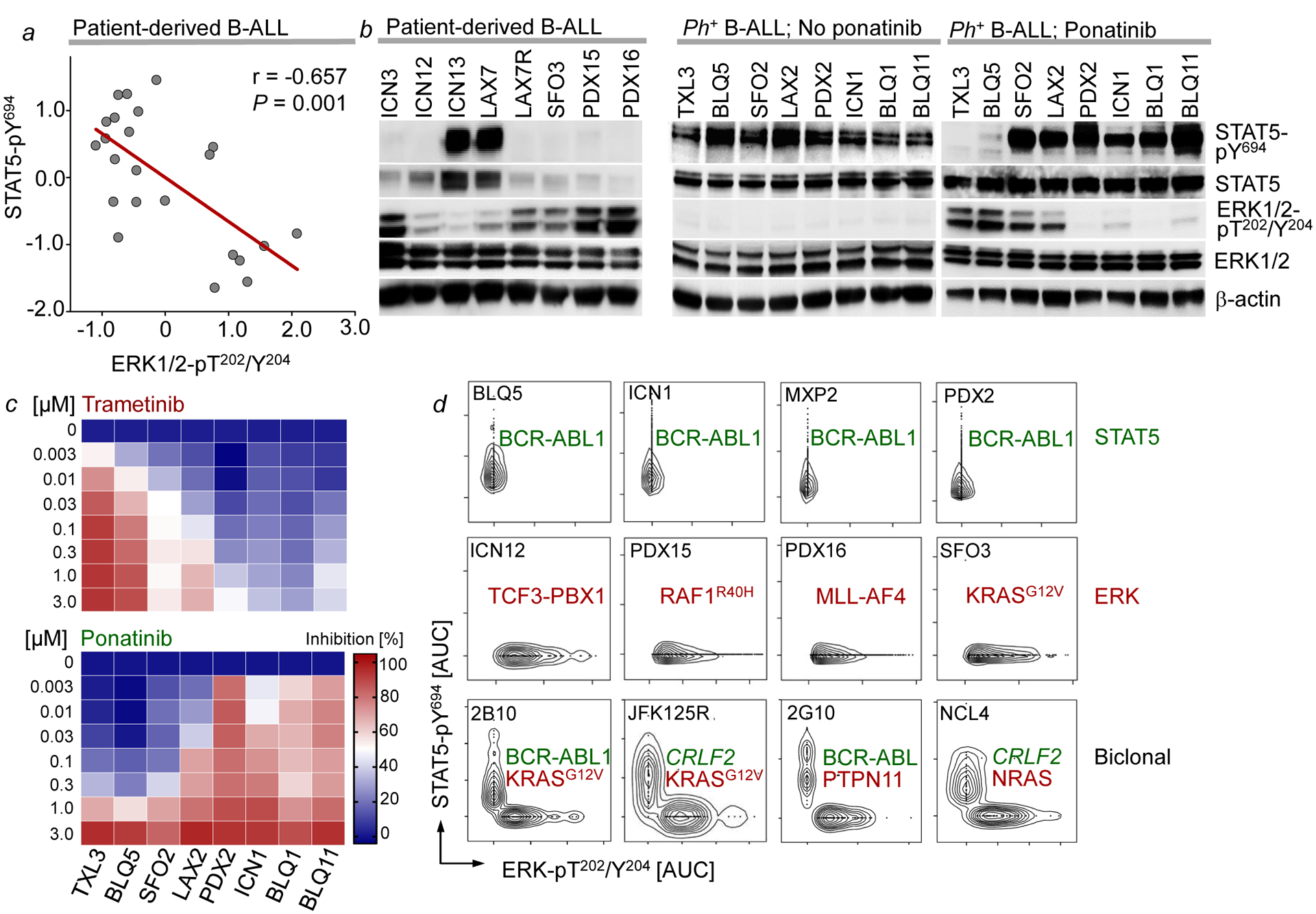
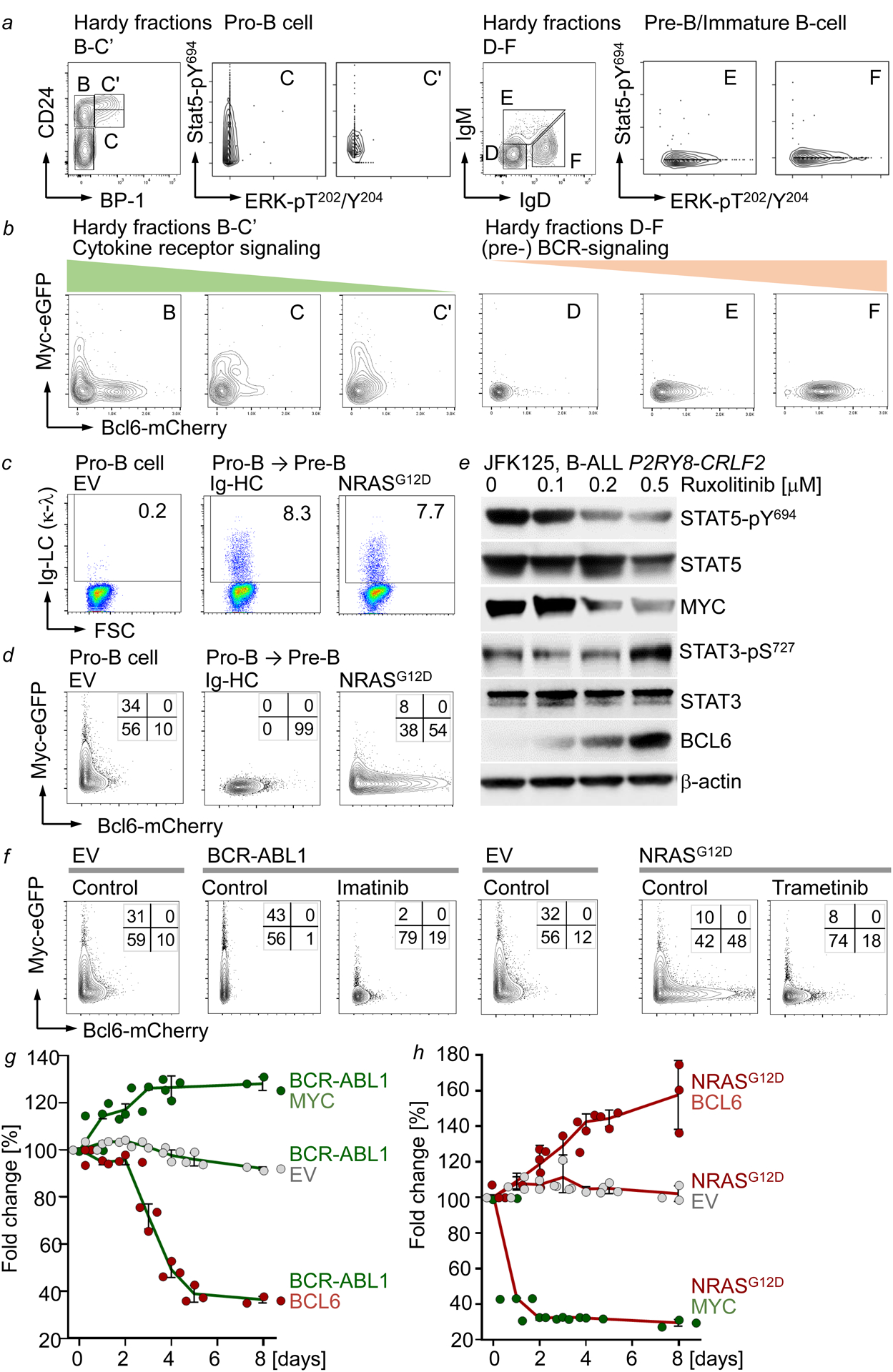
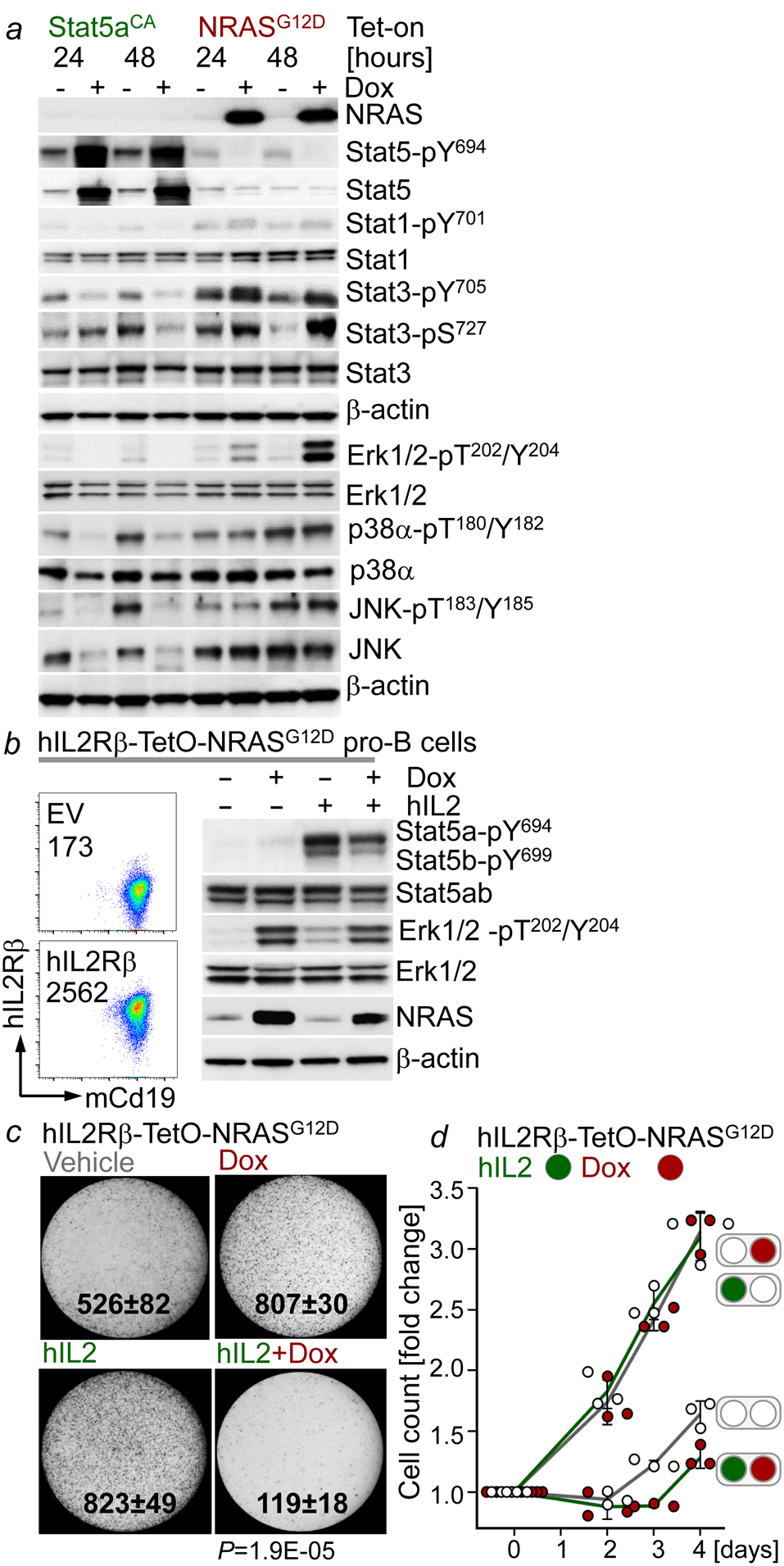
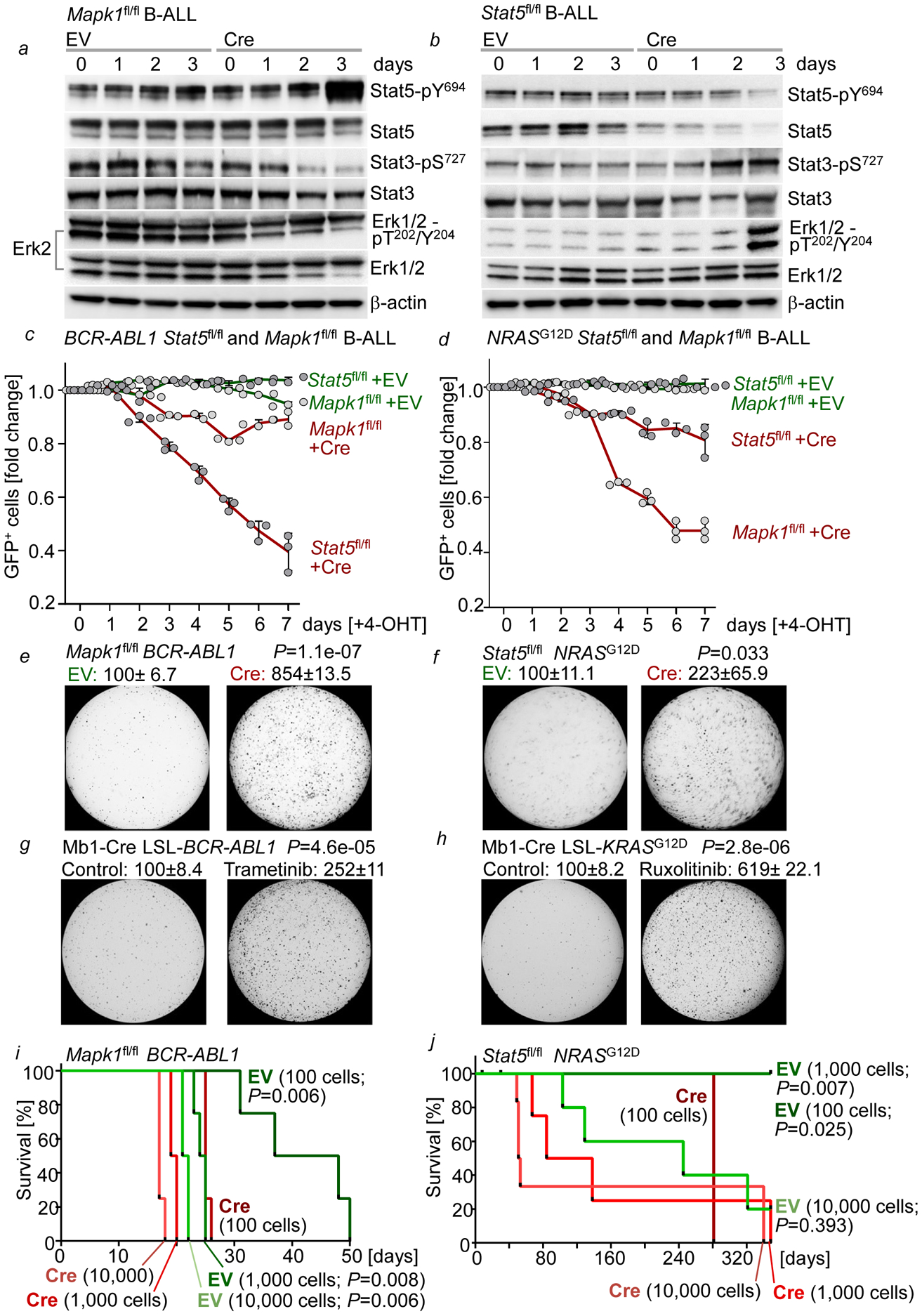
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
