

Exploiting the Therapeutic Interaction of WNT Pathway Activation and Asparaginase for Colorectal Cancer Therapy
- PMID: 32703769
- PMCID: PMC7642035
- DOI: 10.1158/2159-8290.CD-19-1472
Exploiting the Therapeutic Interaction of WNT Pathway Activation and Asparaginase for Colorectal Cancer Therapy
Abstract
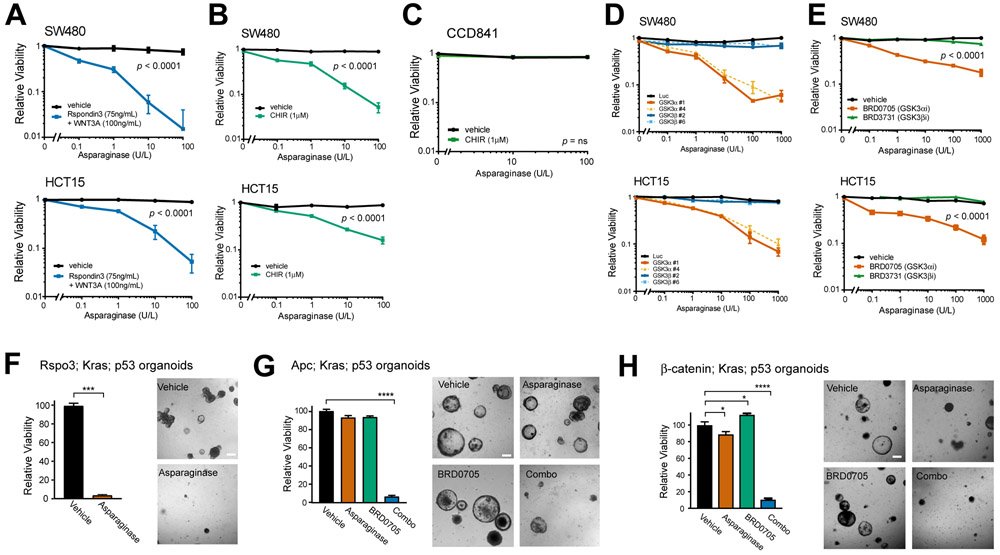
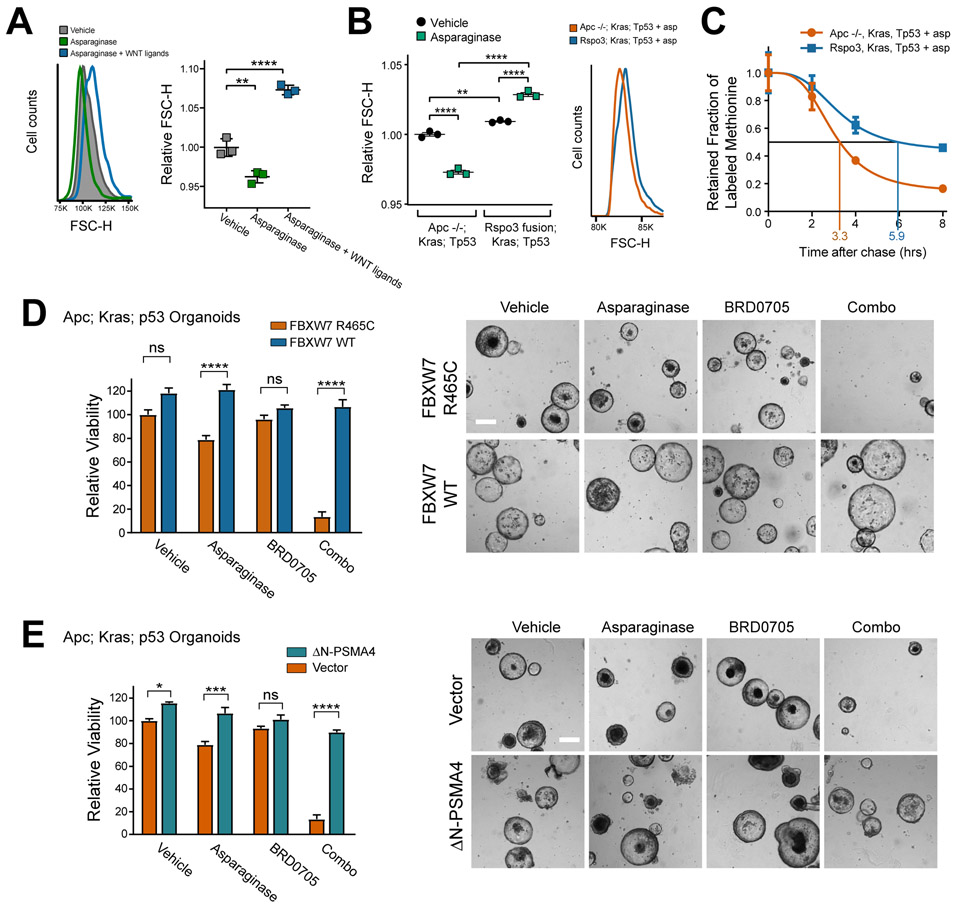
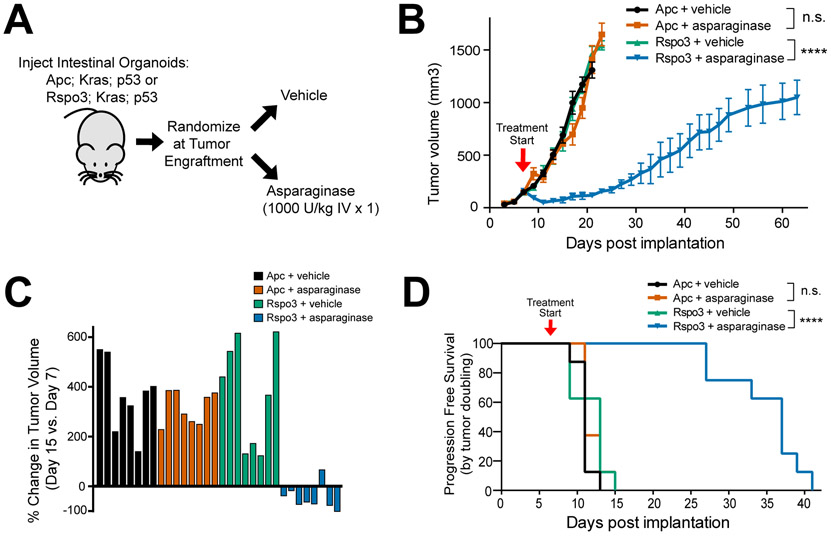
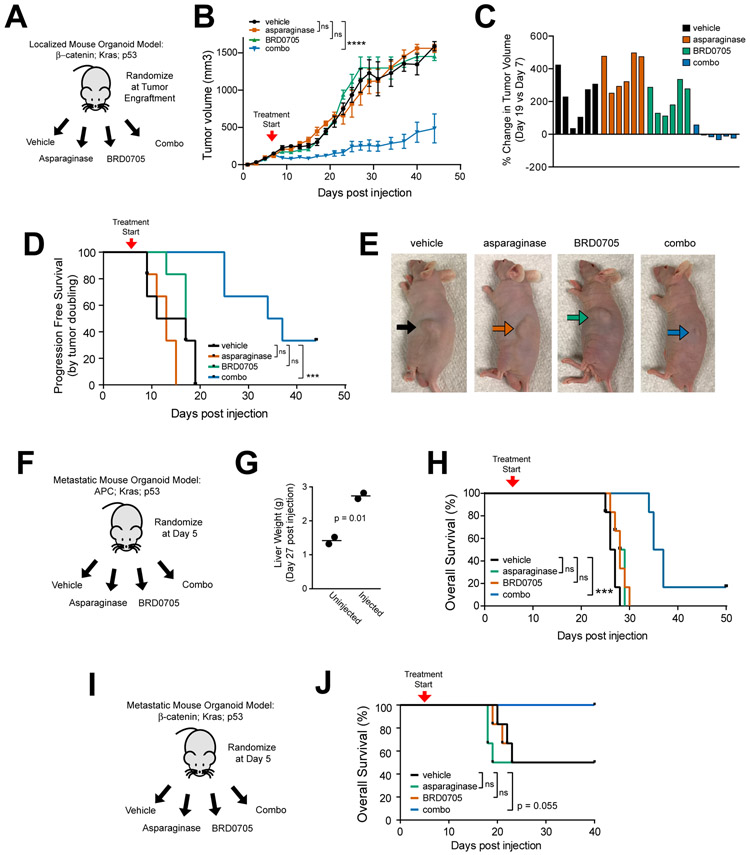
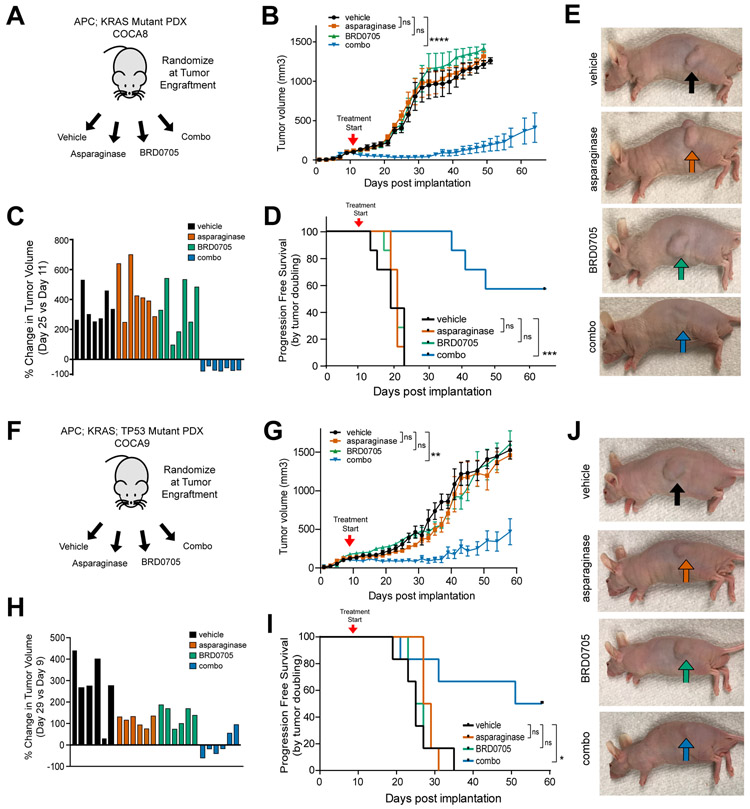
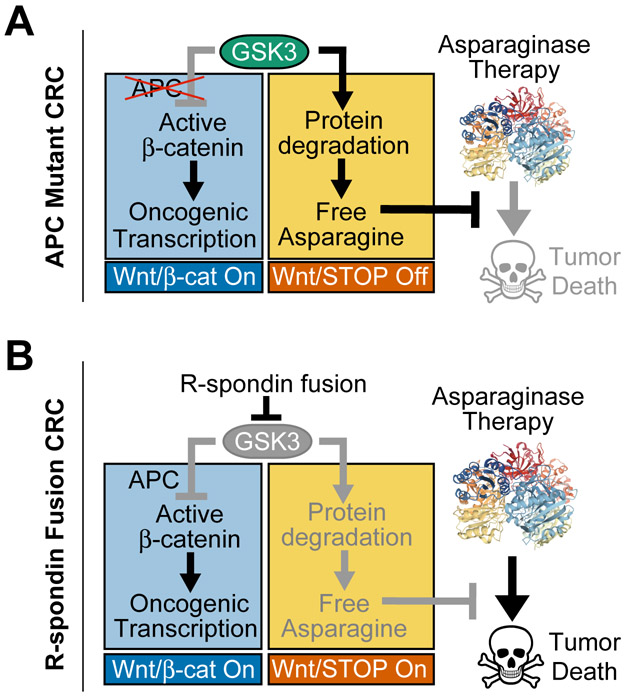
Colorectal cancer is driven by mutations that activate canonical WNT/β-catenin signaling, but inhibiting WNT has significant on-target toxicity, and there are no approved therapies targeting dominant oncogenic drivers. We recently found that activating a β-catenin-independent branch of WNT signaling that inhibits GSK3-dependent protein degradation induces asparaginase sensitivity in drug-resistant leukemias. To test predictions from our model, we turned to colorectal cancer because these cancers can have WNT-activating mutations that function either upstream (i.e., R-spondin fusions) or downstream (APC or β-catenin mutations) of GSK3, thus allowing WNT/β-catenin and WNT-induced asparaginase sensitivity to be unlinked genetically. We found that asparaginase had little efficacy in APC or β-catenin-mutant colorectal cancer, but was profoundly toxic in the setting of R-spondin fusions. Pharmacologic GSK3α inhibition was sufficient for asparaginase sensitization in APC or β-catenin-mutant colorectal cancer, but not in normal intestinal progenitors. Our findings demonstrate that WNT-induced therapeutic vulnerabilities can be exploited for colorectal cancer therapy. SIGNIFICANCE: Solid tumors are thought to be asparaginase-resistant via de novo asparagine synthesis. In leukemia, GSK3α-dependent protein degradation, a catabolic amino acid source, mediates asparaginase resistance. We found that asparaginase is profoundly toxic to colorectal cancers with WNT-activating mutations that inhibit GSK3. Aberrant WNT activation can provide a therapeutic vulnerability in colorectal cancer.See related commentary by Davidsen and Sullivan, p. 1632.This article is highlighted in the In This Issue feature, p. 1611.
©2020 American Association for Cancer Research.
Figures
Comment in
-
Free Asparagine or Die: Cancer Cells Require Proteasomal Protein Breakdown to Survive Asparagine Depletion.Cancer Discov. 2020 Nov;10(11):1632-1634. doi: 10.1158/2159-8290.CD-20-1251. Cancer Discov. 2020. PMID: 33139341
References
-
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992;256(5057):668–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
