

Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy
- PMID: 32703943
- PMCID: PMC7378245
- DOI: 10.1038/s41467-020-17452-6
Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy
Abstract
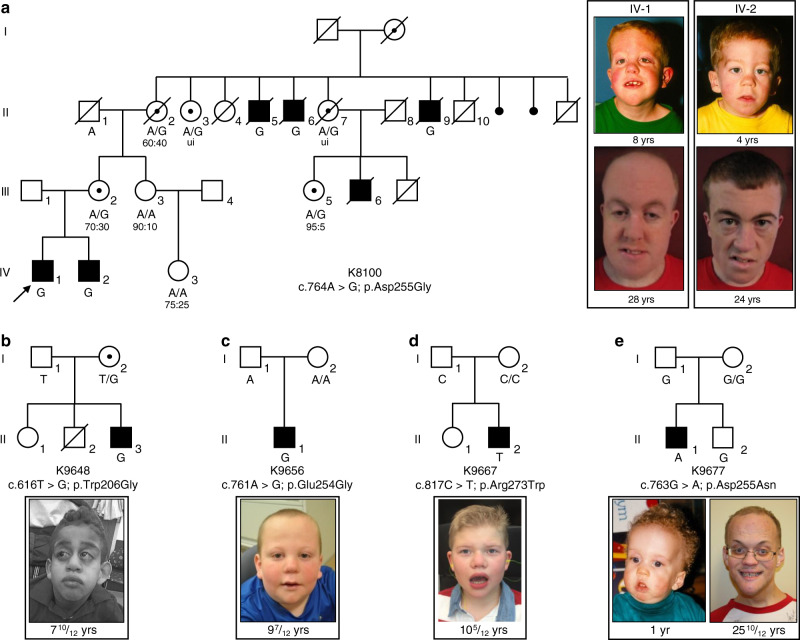
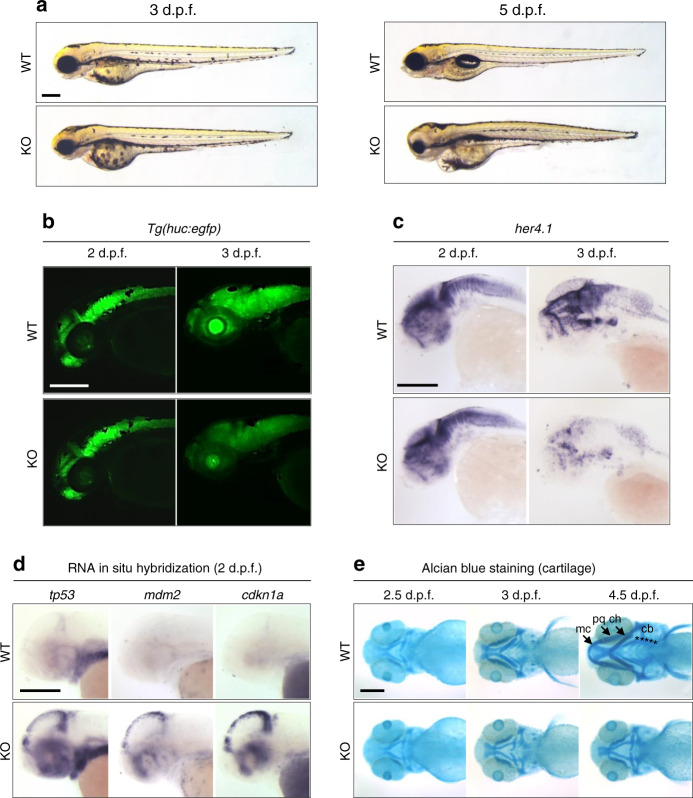
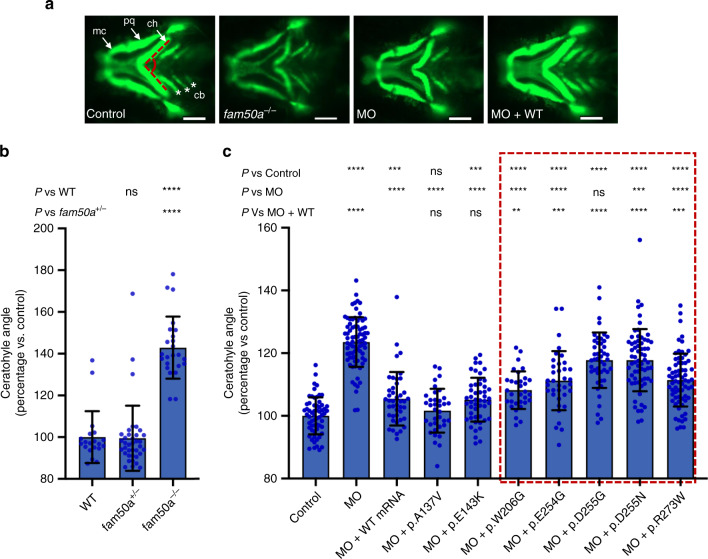
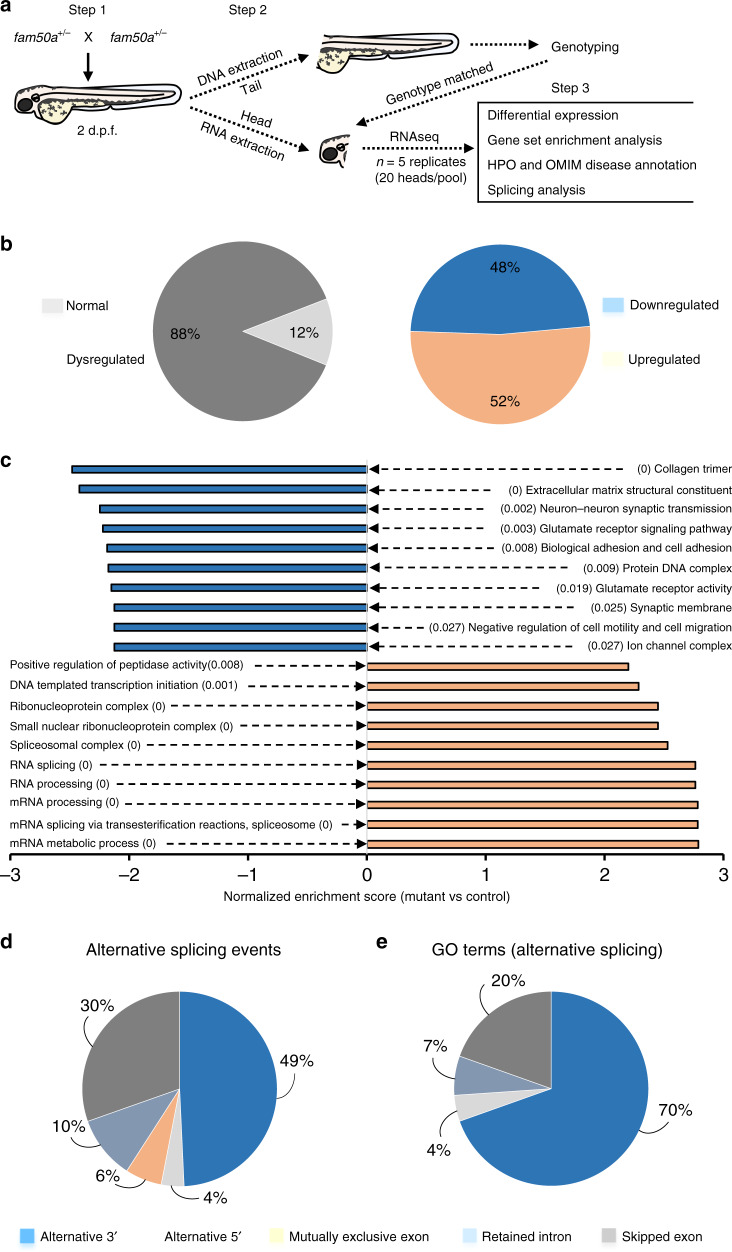
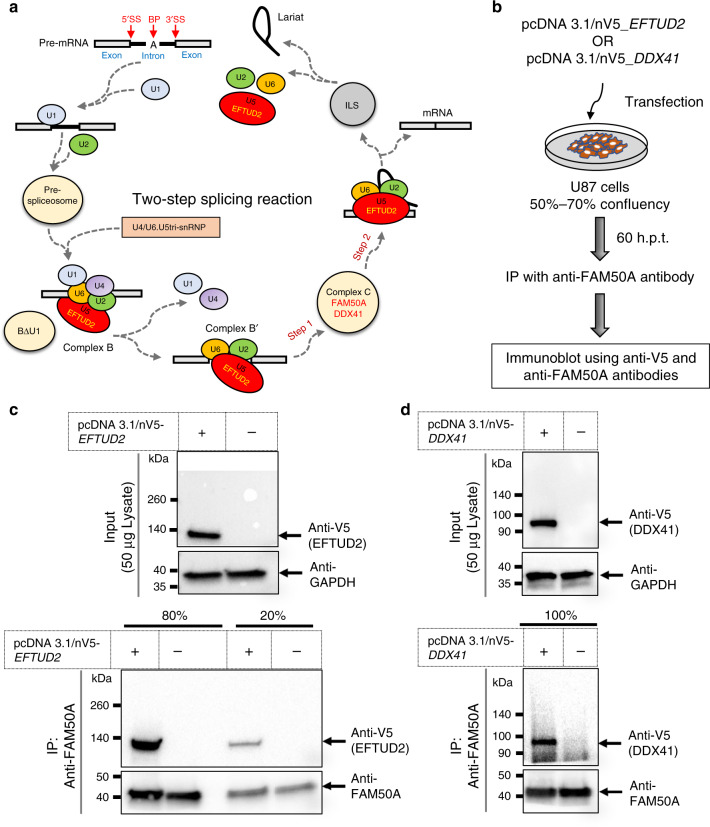
Intellectual disability (ID) is a heterogeneous clinical entity and includes an excess of males who harbor variants on the X-chromosome (XLID). We report rare FAM50A missense variants in the original Armfield XLID syndrome family localized in Xq28 and four additional unrelated males with overlapping features. Our fam50a knockout (KO) zebrafish model exhibits abnormal neurogenesis and craniofacial patterning, and in vivo complementation assays indicate that the patient-derived variants are hypomorphic. RNA sequencing analysis from fam50a KO zebrafish show dysregulation of the transcriptome, with augmented spliceosome mRNAs and depletion of transcripts involved in neurodevelopment. Zebrafish RNA-seq datasets show a preponderance of 3' alternative splicing events in fam50a KO, suggesting a role in the spliceosome C complex. These data are supported with transcriptomic signatures from cell lines derived from affected individuals and FAM50A protein-protein interaction data. In sum, Armfield XLID syndrome is a spliceosomopathy associated with aberrant mRNA processing during development.
Conflict of interest statement
I.W., A.T., and K.M. are employees of GeneDx, Inc. N.K. is a shareholder in Rescindo Therapeutics. The remaining authors declare no competing interests.
Figures

References
-
- Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev. Disabil. 2011;32:419–436. - PubMed
-
- Armfield K, et al. X-linked mental retardation syndrome with short stature, small hands and feet, seizures, cleft palate, and glaucoma is linked to Xq28. Am. J. Med. Genet. 1999;85:236–242. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
