

Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants
- PMID: 32705143
- PMCID: PMC7447512
- DOI: 10.1093/brain/awaa178
Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants
Abstract
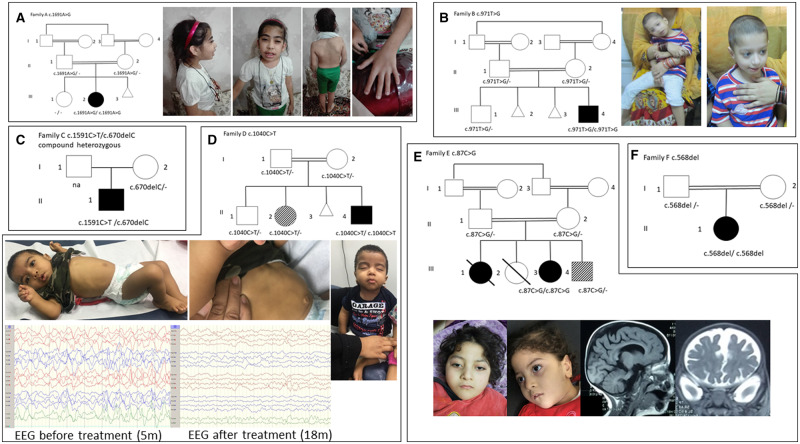
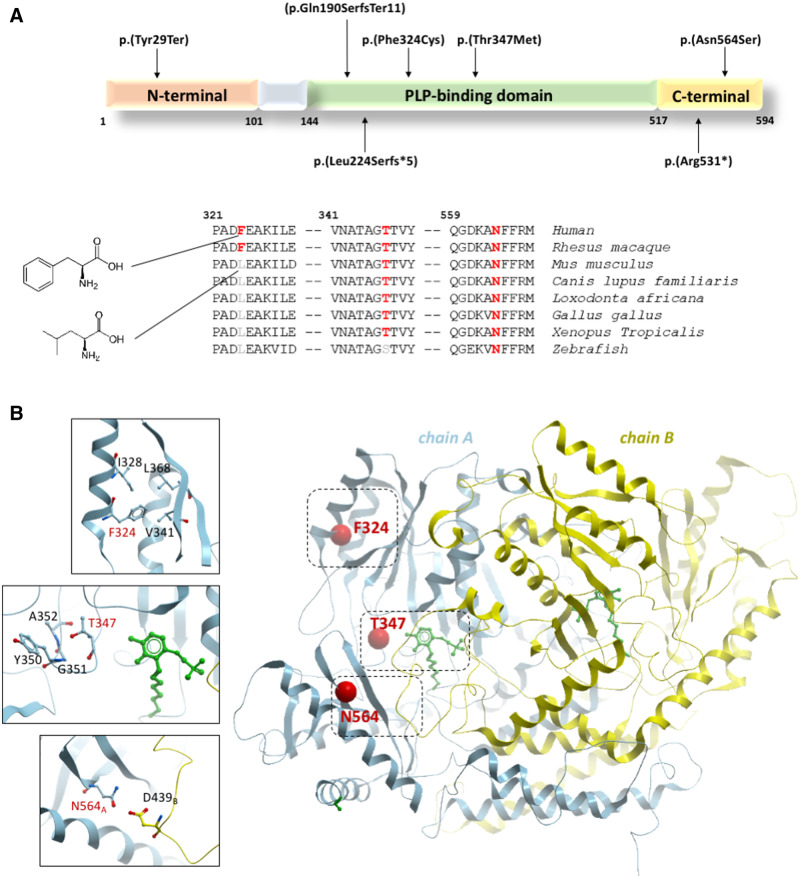
Gamma-aminobutyric acid (GABA) and glutamate are the most abundant amino acid neurotransmitters in the brain. GABA, an inhibitory neurotransmitter, is synthesized by glutamic acid decarboxylase (GAD). Its predominant isoform GAD67, contributes up to ∼90% of base-level GABA in the CNS, and is encoded by the GAD1 gene. Disruption of GAD1 results in an imbalance of inhibitory and excitatory neurotransmitters, and as Gad1-/- mice die neonatally of severe cleft palate, it has not been possible to determine any potential neurological dysfunction. Furthermore, little is known about the consequence of GAD1 disruption in humans. Here we present six affected individuals from six unrelated families, carrying bi-allelic GAD1 variants, presenting with developmental and epileptic encephalopathy, characterized by early-infantile onset epilepsy and hypotonia with additional variable non-CNS manifestations such as skeletal abnormalities, dysmorphic features and cleft palate. Our findings highlight an important role for GAD1 in seizure induction, neuronal and extraneuronal development, and introduce GAD1 as a new gene associated with developmental and epileptic encephalopathy.
Keywords: GAD1; cleft palate; epilepsy; muscle weakness; neurodevelopmental delay.
© The Author(s) (2020). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
References
-
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, et al.Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun 1996; 229: 891–895. - PubMed
-
- Berecki G, Bryson A, Terhag J, Maljevic S, Gazina EV, Hill SL, et al.SCN1A gain of function in early infantile encephalopathy. Ann Neurol 2019; 85: 514–25. - PubMed
-
- Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P.. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol 2010; 14: 146–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G1001253/MRC_/Medical Research Council/United Kingdom
- R605/0717/DMT_/The Dunhill Medical Trust/United Kingdom
- G108/638/MRC_/Medical Research Council/United Kingdom
- WT104033AIA/WT_/Wellcome Trust/United Kingdom
- WT093205/WT_/Wellcome Trust/United Kingdom
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- G0802760/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- MC_UP_1502/3/MRC_/Medical Research Council/United Kingdom
- MR/S005021/1/MRC_/Medical Research Council/United Kingdom
- G0601943/MRC_/Medical Research Council/United Kingdom
- MR/S01165X/1/MRC_/Medical Research Council/United Kingdom
