

Timeless couples G-quadruplex detection with processing by DDX11 helicase during DNA replication
- PMID: 32705708
- PMCID: PMC7506991
- DOI: 10.15252/embj.2019104185
Timeless couples G-quadruplex detection with processing by DDX11 helicase during DNA replication
Abstract
Regions of the genome with the potential to form secondary DNA structures pose a frequent and significant impediment to DNA replication and must be actively managed in order to preserve genetic and epigenetic integrity. How the replisome detects and responds to secondary structures is poorly understood. Here, we show that a core component of the fork protection complex in the eukaryotic replisome, Timeless, harbours in its C-terminal region a previously unappreciated DNA-binding domain that exhibits specific binding to G-quadruplex (G4) DNA structures. We show that this domain contributes to maintaining processive replication through G4-forming sequences, and exhibits partial redundancy with an adjacent PARP-binding domain. Further, this function of Timeless requires interaction with and activity of the helicase DDX11. Loss of both Timeless and DDX11 causes epigenetic instability at G4-forming sequences and DNA damage. Our findings indicate that Timeless contributes to the ability of the replisome to sense replication-hindering G4 formation and ensures the prompt resolution of these structures by DDX11 to maintain processive DNA synthesis.
Keywords: DNA replication; G-quadruplex; fork protection complex; replisome; timeless.
© 2020 MRC Laboratory of Molecular Biology. Published under the terms of the CC BY 4.0 license.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
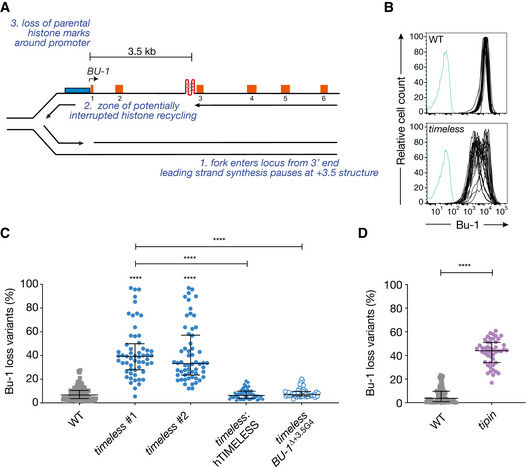
The BU‐1 locus as a model system to record G4‐dependent replication stalling. The leading strand of a replication fork entering the locus from the 3′ end stochastically stalls at the +3.5 G4, leading to the formation of a region of ssDNA, with interruption of parental histone recycling and of histone modifications necessary to maintain normal expression of the locus (Schiavone et al, 2014).
Instability of BU‐1 expression in timeless cells. FACS plots of wild‐type and timeless (clone 1) DT40 cells stained with anti‐Bu-1 conjugated with phycoerythrin. Each line represents the Bu‐1 expression profile of an individual clonal population. Unstained controls are shown in blue.
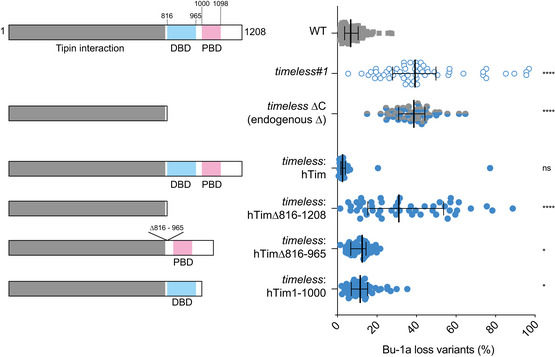
Fluctuation analysis for Bu‐1 loss in wild‐type DT40 cells and two independent timeless clones generated by CRISPR‐Cas9 targeting (clones 1 and 2; Appendix Fig S1), timeless (clone 1) complemented by expression of human Timeless cDNA and a timeless mutant on a background in which the endogenous +3.5 G4 has been deleted (ΔG4) (Schiavone et al, 2014).
Fluctuation analysis for Bu‐1 loss in DT40 wild‐type and tipin cells.
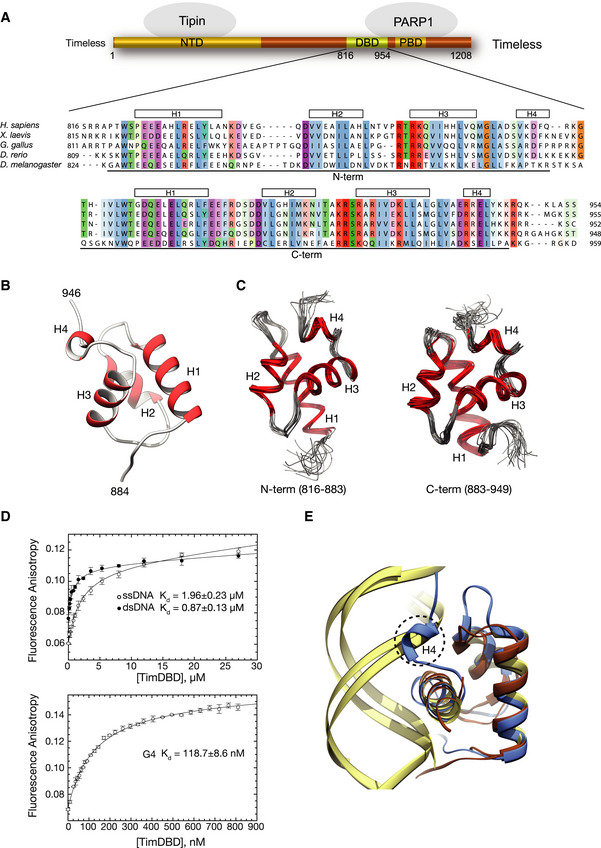
Schematic drawing of human Timeless and its known domain structure (NTD: N‐terminal domain; DBD: DNA‐binding domain; PBD: PARP‐binding domain). A multiple sequence alignment of vertebrate Timeless sequences is shown underneath, with amino acid conservation coloured according to the Clustal colour scheme. The alignment is annotated with the extent and secondary structure elements of the two helical domains (N‐term and C‐term) composing the DBD.
Ribbon drawing of the 1.15 Å crystal structure at of the DBD C‐term. Helices are in red and labelled H1–H4.
Ribbon drawings of the N‐term and C‐term domains of the DBD determined by NMR. The two domains are shown in the same orientation to highlight their high degree of three‐dimensional similarity. The superposition of the 20 lowest energy structures is shown for each domain.
The DNA‐binding affinity of DBD was measured by fluorescence anisotropy, titrating the DBD protein against Cy3 3′‐labelled ssDNA, dsDNA and G4 DNA (see Appendix Table S2 for sequence details). The top panel shows binding curves for ss‐ and dsDNA, and the bottom panel shows the binding curve for the G4 DNA substrate. The data points represent the mean of at least three independent experiments, and the error bars indicate one standard deviation (SD).
Ribbon diagram of the superposition of DBD C‐term with the highly similar DNA‐binding domains of telomeric protein TRF1 (PDB ID 1W0T) (Court et al, 2005) and the bacterial cell cycle regulator GcrA (PDB ID 5Z7I) (Wu et al, 2018) in complex with their DNA substrates. A similar DNA‐binding mode by DBD would cause a steric overlap of helix H4 with the phosphate backbone of dsDNA. DBD C‐term is in light blue, TRF1 and GcrA proteins in brown and their DNA substrates in khaki.

ssG4: G4 flanked by single‐stranded DNA; ssHP: hairpin flanked by single‐stranded DNA; ss: single‐stranded DNA (Appendix Table S2 for sequence details).
Binding affinity of Timeless–Tipin for a range of G4 DNA sequences (see Appendix Table S2 for sequence details and references). Single‐stranded (ss20: 5′‐6FAM-ATAAGAGTGGTTAGAGTGTA) and double‐stranded (ds20: ss20 annealed to complementary sequence) DNA were also tested as controls.
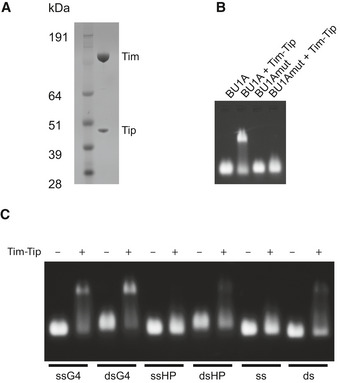
Coomassie‐stained SDS–PAGE gel of purified Timeless–Tipin complex.
Electrophoretic mobility shift assay (EMSA) showing the binding of Timeless–Tipin to G‐quadruplex sequence BU1A + 3.5. Mutation of the G‐quadruplex sequence (BU1A + 3.5 mut) disrupts Timeless–Tipin binding (see Appendix Table S2 for sequence details). Timeless–Tipin and DNA are both present at a final concentration of 5 μM.
EMSA showing the binding of Timeless–Tipin to G‐quadruplex sequences (ssG4, dsG4) but not single‐stranded DNA (ss), double‐stranded DNA (ds) or hairpin‐containing sequences (ssHP, dsHP). Timeless–Tipin and DNA are both present at a final concentration of 5 μM.

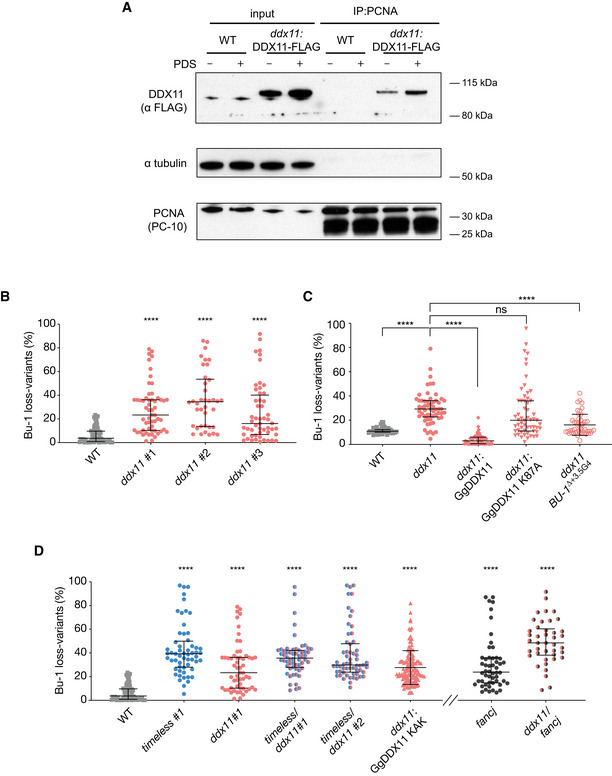
Enhanced recruitment of DDX11 to chromatin associated PCNA following exposure to 4 μM PDS for 24 h. PCNA was precipitated from cross‐linked chromatin and the immunoprecipitate blotted for FLAG‐DDX11.
Fluctuation analysis for Bu‐1a loss in wild‐type DT40 cells, two independent ddx11 clones generated by CRISPR‐Cas9 targeting (clones 1 and 2) and one ddx11 clone generated by conventional homologous recombination gene targeting (clone 3). Each symbol represents the percentage of cells in an individual clone expanded for 2–3 weeks that have lost Bu‐1ahigh expression.
Fluctuation analysis for Bu‐1a loss variant generation in wild‐type cells, ddx11 (clone 1) cells, ddx11 (clone 1) complemented by expression of chicken DDX11 WT cDNA, ddx11 (clone 1) complemented by expression of helicase‐dead form of chicken DDX11 (K87A) cDNA, and a ddx11 clone generated in cells in which the endogenous +3.5 G4 has been deleted (ΔG4).
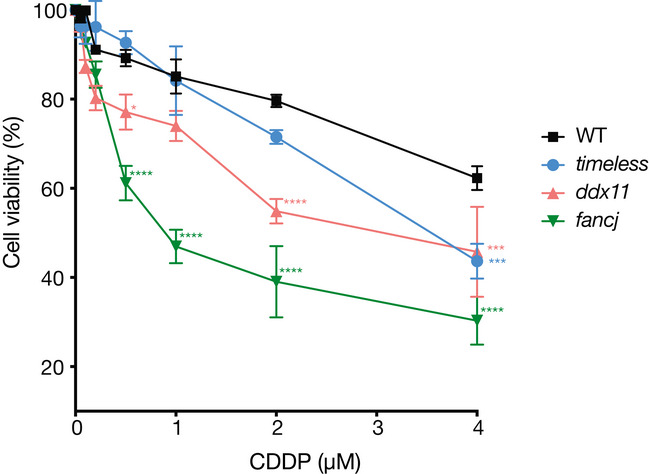
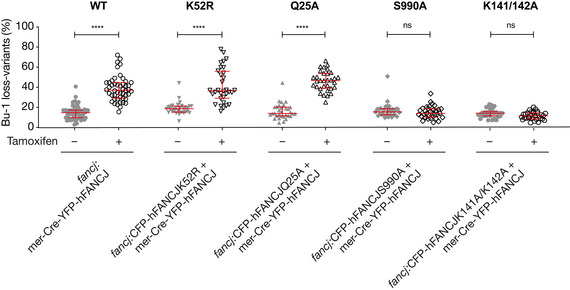
Fluctuation analysis for Bu‐1 loss in two independent timeless/ddx11 double‐mutant clones (#1 and #2), ddx11 expressing DDX11KAK (see Appendix Fig S3), and fancj and fancj/ddx11 double mutants. Fluctuation analyses for wild type, timeless #1 (Fig 44) and ddx11 #1 (Fig 55A) are shown for comparison.
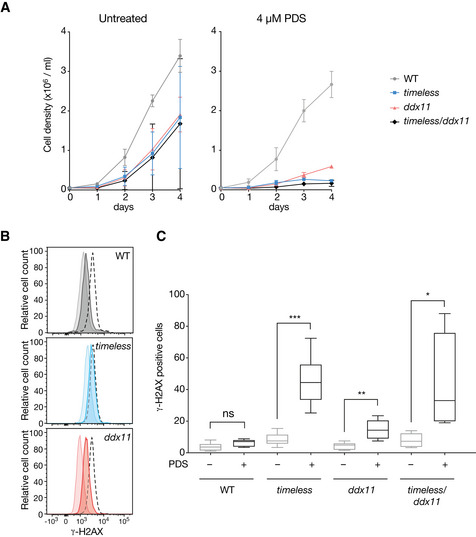
Growth curves for DT40 wild type, ddx11, timeless and ddx11/timeless cells, with and without 4 μM pyridostatin (PDS). Cells were seeded at 5 × 104 cells/ml on day 0 and the viable cells were counted each 24 h for 4 days. Bars represent SD of two independent experiments performed in duplicate. Doubling times (DMSO): WT 13 h, timeless 18 h, ddx11 16 h, ddx11/timeless 24 h. Doubling times (PDS): WT 13.6 h, timeless 27 h, ddx11 25.7 h, ddx11/timeless 47.5 h.
DDR signalling detected by phosphorylation of histone H2AX (γ‐H2AX) by flow cytometry in untreated cells or cells exposed to 4 μM PDS for 3 days. Pale histogram, untreated; dark histogram, treated; black dotted line, positive control cells treated with 0.1 μM cisplatin, also for 3 days.
Quantification of γ‐H2AX in DT40 wild type, ddx11, timeless and ddx11/timeless cells treated with 4 μM PDS for 3 days. The central band represents the median, the box the 25th–75th centile and whiskers the minimum to maximum range of three independent experiments performed in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001 unpaired, two‐tailed t‐test for each pairwise comparison ± PDS. See also Appendix Fig S4 for immunofluorescence images of the γ‐H2AX signal.
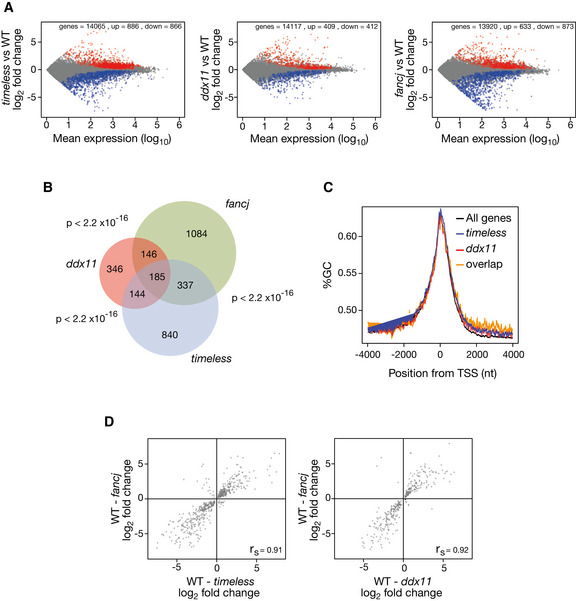
Dysregulated genes in timeless (left panel), ddx11 (centre panel) and fancj (right panel) mutants relative to wild type. All genes with > 1 transcript per million in both conditions are plotted. Significantly (P ≥ 0.95) upregulated genes shown in red; downregulated in blue.
Venn diagram showing the overlap in genes deregulated in timeless, ddx11 and fancj relative to wild type. P < 2.2 × 10−16 for each pairwise comparison (Fisher hypergeometric distribution).
GC content around the TSS in genes dysregulated in timeless (blue), ddx11 (red) and in both mutants (“overlap”, orange) compared with all genes (black).
Correlation of magnitude and direction of change of genes dysregulated (relative to wild type) in fancj vs. timeless (left panel) and ddx11 (right panel) DT40 cells. r s (Spearman rho) is shown for each correlation.
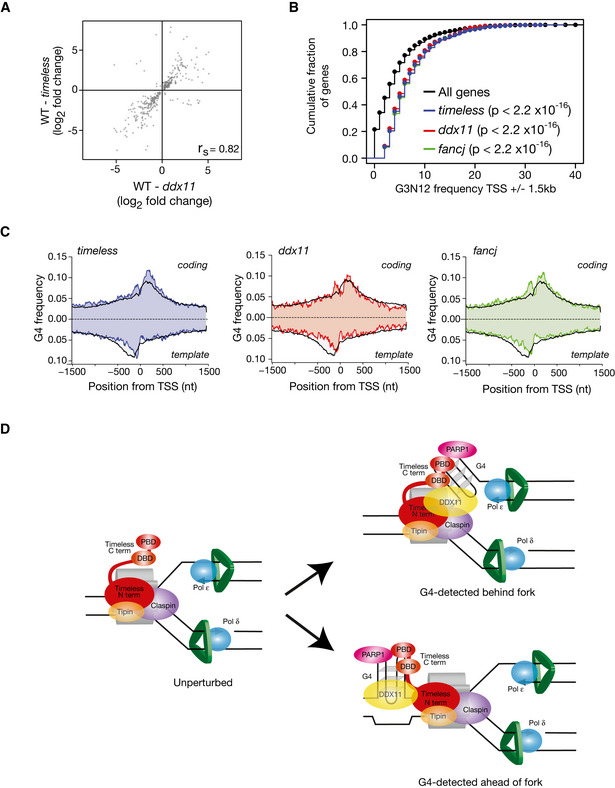
Correlation of magnitude and direction of change of genes dysregulated (relative to wild type) in timeless vs. ddx11 DT40 cells. r s (Spearman rho) is shown for each correlation.
Genes dysregulated in timeless, ddx11 and fancj mutants have a higher density of G4s around their TSS. Cumulative fraction of the genes dysregulated in timeless (red), ddx11 (blue) and fancj (green) containing n (x‐axis) G4 motifs within 1.5 kb of the TSS compared with all genes (black). P values calculated with the Kolmogorov–Smirnov test.
Metagene analysis showing G4 frequency around the TSS of genes dysregulated (up or down) in timeless (left panel), ddx11 (centre panel) and fancj (right panel) compared with all genes (black line). G4 frequency is calculated separately for coding (above the x‐axis) and template strands (below the x‐axis).
A model for recognition and processing of replisome associated G4s by Timeless/DDX11. Current evidence suggests that Timeless is a constitutive component of the replication fork. We suggest that the C‐terminus of Timeless may help detect G4s in the vicinity of the replisome by a combination of direct recognition through the DNA‐binding domain and indirectly through the PARP‐binding domain. It is not currently possible to distinguish whether this mechanism would operate ahead or behind the fork itself (Lerner & Sale, 2019), although recent structural evidence placing Tof1, the yeast homologue of Timeless, ahead of the fork (Baretić et al, 2020) would appear to make the first possibility more probable. However, for failure to resolve G4s ahead of the fork to result in uncoupling would require the CMG helicase to traverse the structure, as has been suggested for interstrand crosslinks (Sparks et al, 2019).
Comment in
-
A Timeless Tale: G4 structure recognition by the fork protection complex triggers unwinding by DDX11 helicase.EMBO J. 2020 Sep 15;39(18):e106305. doi: 10.15252/embj.2020106305. Epub 2020 Aug 13. EMBO J. 2020. PMID: 32790898 Free PMC article.
References
-
- Ambrus A, Chen D, Dai J, Jones RA, Yang D (2005) Solution structure of the biologically relevant G‐quadruplex element in the human c‐MYC promoter. Implications for G‐quadruplex stabilization. Biochemistry 44: 2048–2058 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
