

Whole-genome analyses reveal gene content differences between nontypeable Haemophilus influenzae isolates from chronic obstructive pulmonary disease compared to other clinical phenotypes
- PMID: 32706329
- PMCID: PMC7641420
- DOI: 10.1099/mgen.0.000405
Whole-genome analyses reveal gene content differences between nontypeable Haemophilus influenzae isolates from chronic obstructive pulmonary disease compared to other clinical phenotypes
Erratum in
-
Corrigendum: Whole-genome analyses reveal gene content differences between nontypeable Haemophilus influenzae isolates from chronic obstructive pulmonary disease compared to other clinical phenotypes.Microb Genom. 2021 Oct;7(10):000665. doi: 10.1099/mgen.0.000665. Microb Genom. 2021. PMID: 34605761 Free PMC article. No abstract available.
Abstract
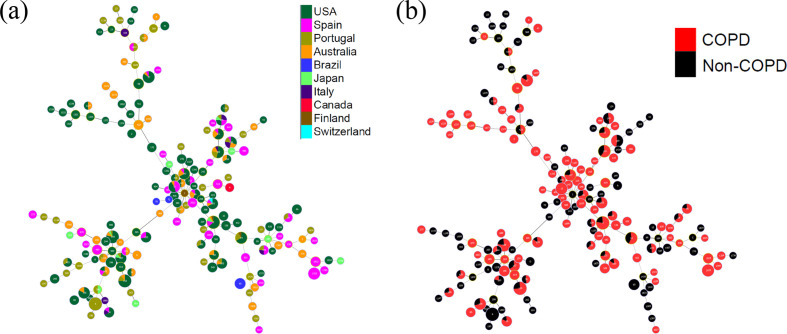
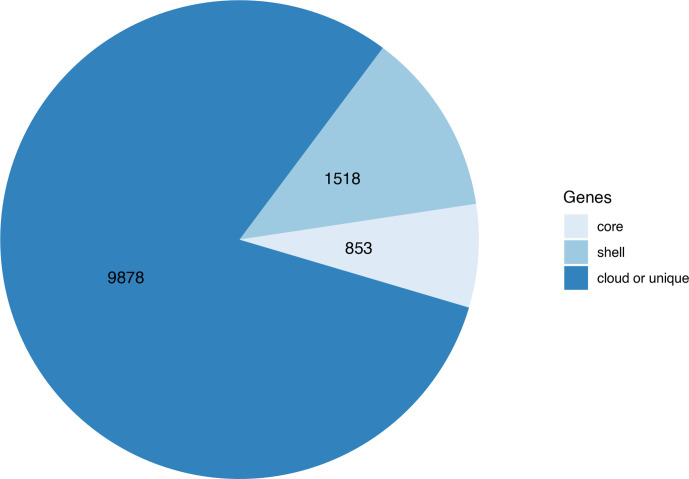
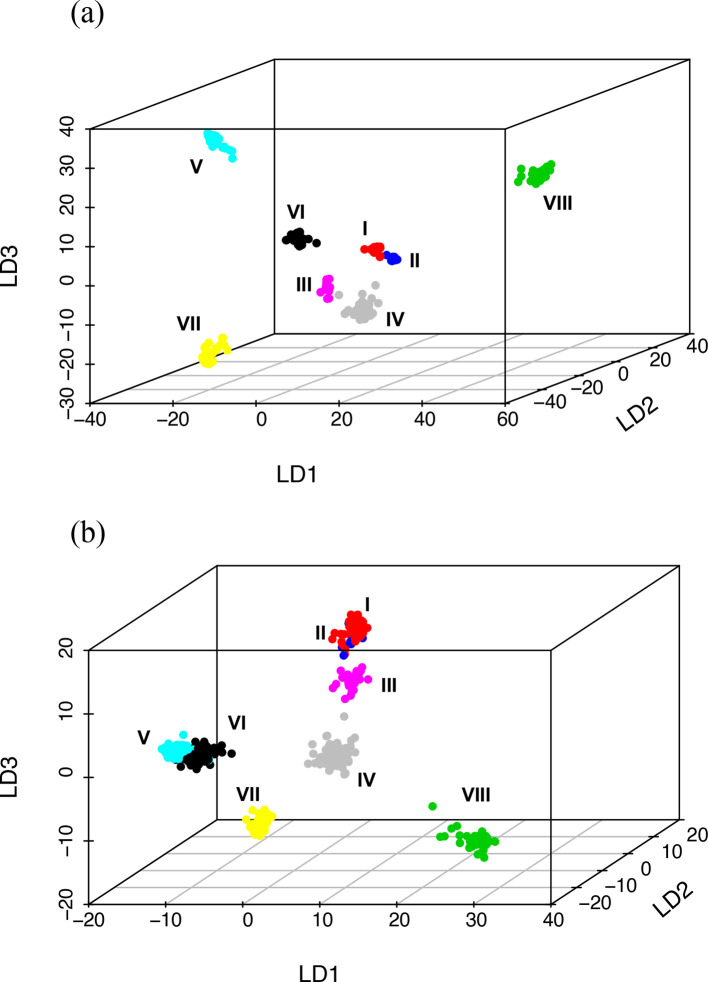
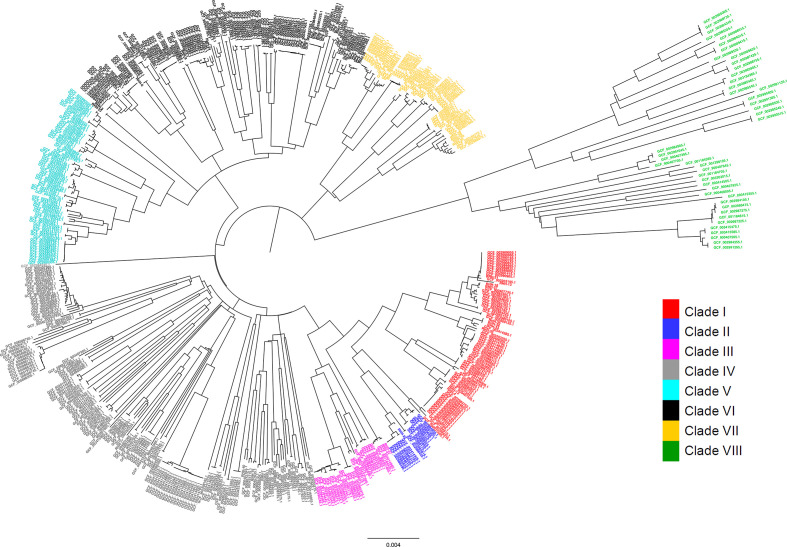
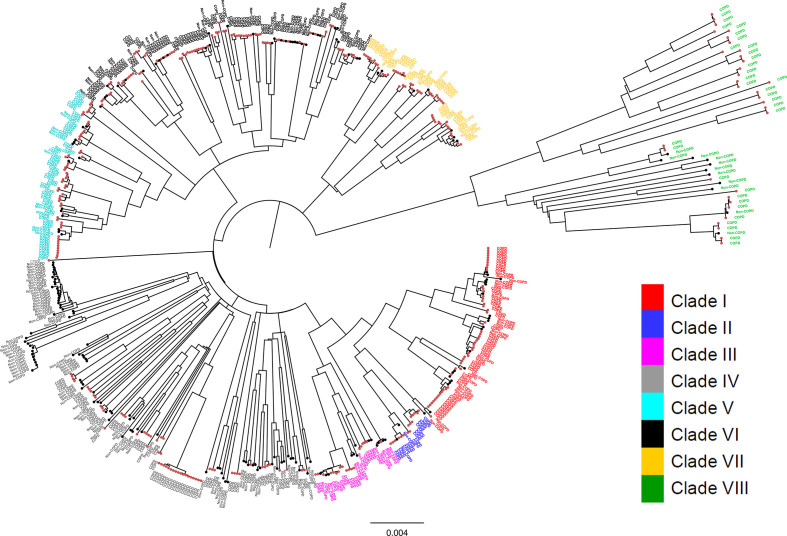
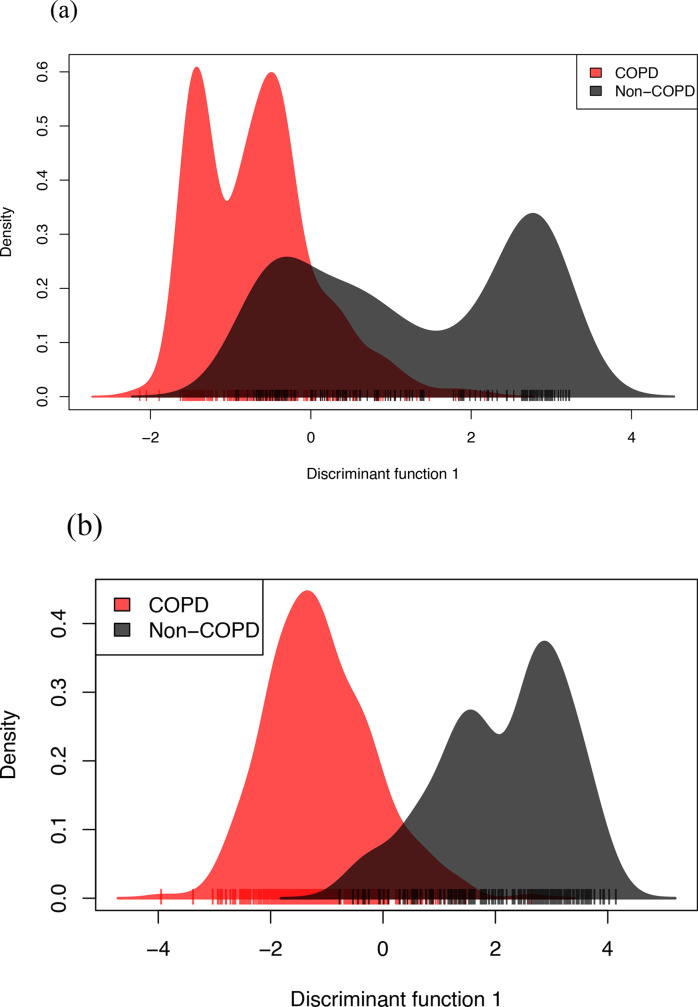
Nontypeable Haemophilus influenzae (NTHi) colonizes human upper respiratory airways and plays a key role in the course and pathogenesis of acute exacerbations of chronic obstructive pulmonary disease (COPD). Currently, it is not possible to distinguish COPD isolates of NTHi from other clinical isolates of NTHi using conventional genotyping methods. Here, we analysed the core and accessory genome of 568 NTHi isolates, including 40 newly sequenced isolates, to look for genetic distinctions between NTHi isolates from COPD with respect to other illnesses, including otitis media, meningitis and pneumonia. Phylogenies based on polymorphic sites in the core-genome did not show discrimination between NTHi strains collected from different clinical phenotypes. However, pan-genome-wide association studies identified 79 unique NTHi accessory genes that were significantly associated with COPD. Furthermore, many of the COPD-related NTHi genes have known or predicted roles in virulence, transmembrane transport of metal ions and nutrients, cellular respiration and maintenance of redox homeostasis. This indicates that specific genes may be required by NTHi for its survival or virulence in the COPD lung. These results advance our understanding of the pathogenesis of NTHi infection in COPD lungs.
Keywords: chronic obstructive pulmonary disease; nontypeable Haemophilus influenzae; pan-genome-wide association studies; whole-genome sequencing.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical