

Kinase Interaction Network Expands Functional and Disease Roles of Human Kinases
- PMID: 32707033
- PMCID: PMC7427327
- DOI: 10.1016/j.molcel.2020.07.001
Kinase Interaction Network Expands Functional and Disease Roles of Human Kinases
Abstract
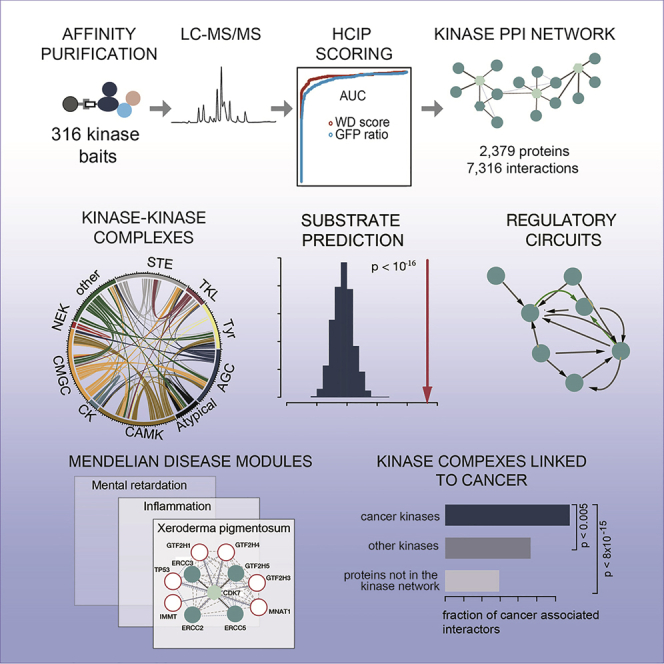
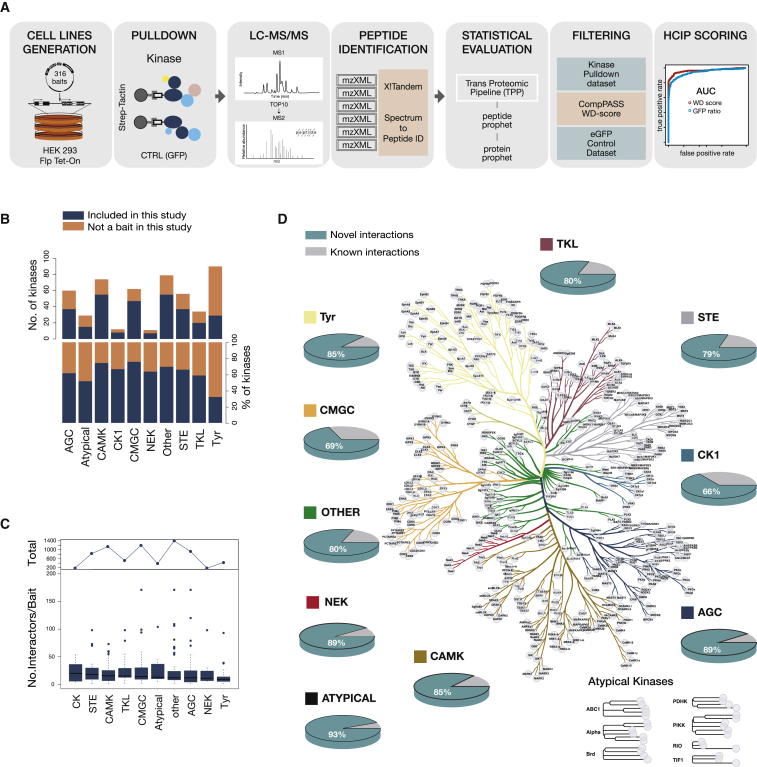
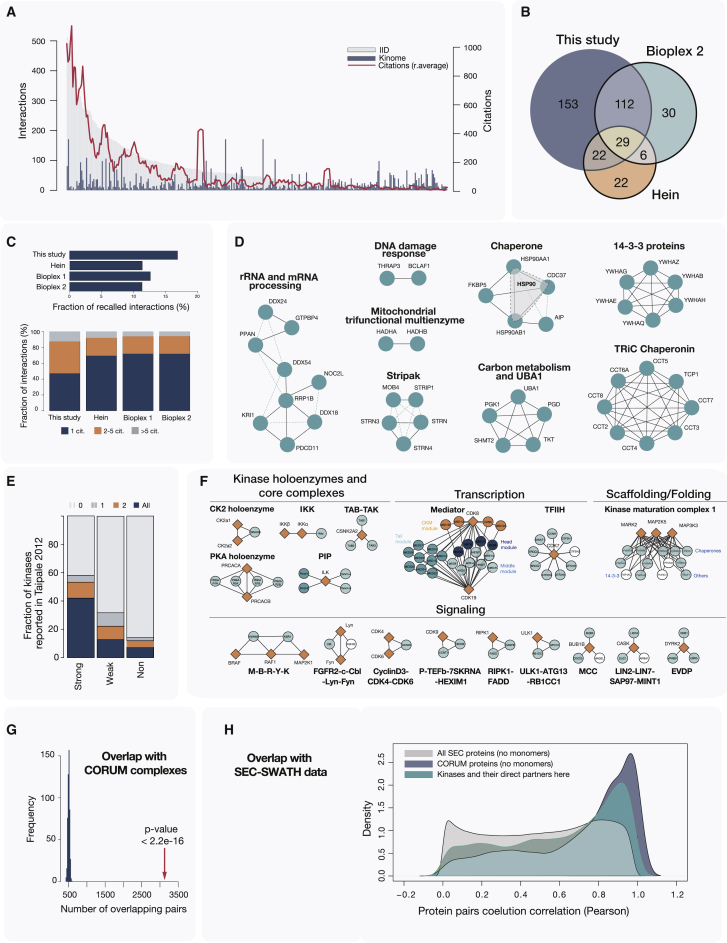
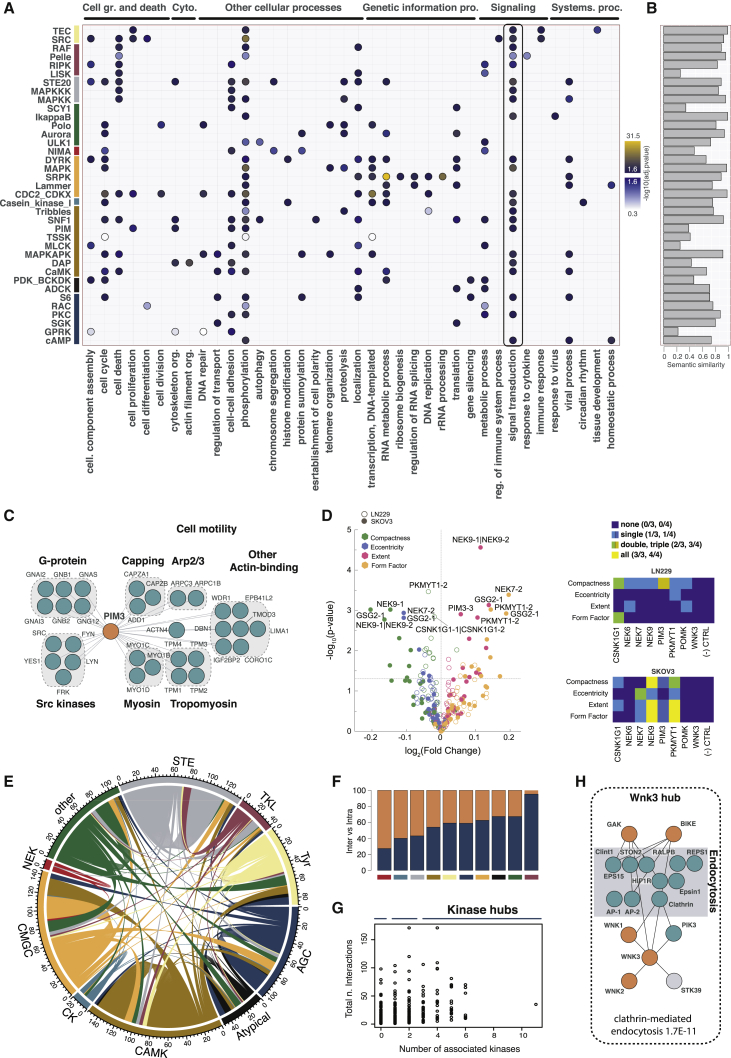
Protein kinases are essential for signal transduction and control of most cellular processes, including metabolism, membrane transport, motility, and cell cycle. Despite the critical role of kinases in cells and their strong association with diseases, good coverage of their interactions is available for only a fraction of the 535 human kinases. Here, we present a comprehensive mass-spectrometry-based analysis of a human kinase interaction network covering more than 300 kinases. The interaction dataset is a high-quality resource with more than 5,000 previously unreported interactions. We extensively characterized the obtained network and were able to identify previously described, as well as predict new, kinase functional associations, including those of the less well-studied kinases PIM3 and protein O-mannose kinase (POMK). Importantly, the presented interaction map is a valuable resource for assisting biomedical studies. We uncover dozens of kinase-disease associations spanning from genetic disorders to complex diseases, including cancer.
Keywords: cancer; disease module; interaction network; kinome; protein complexes; protein kinases; proteomics; signaling regulation; systems biology.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
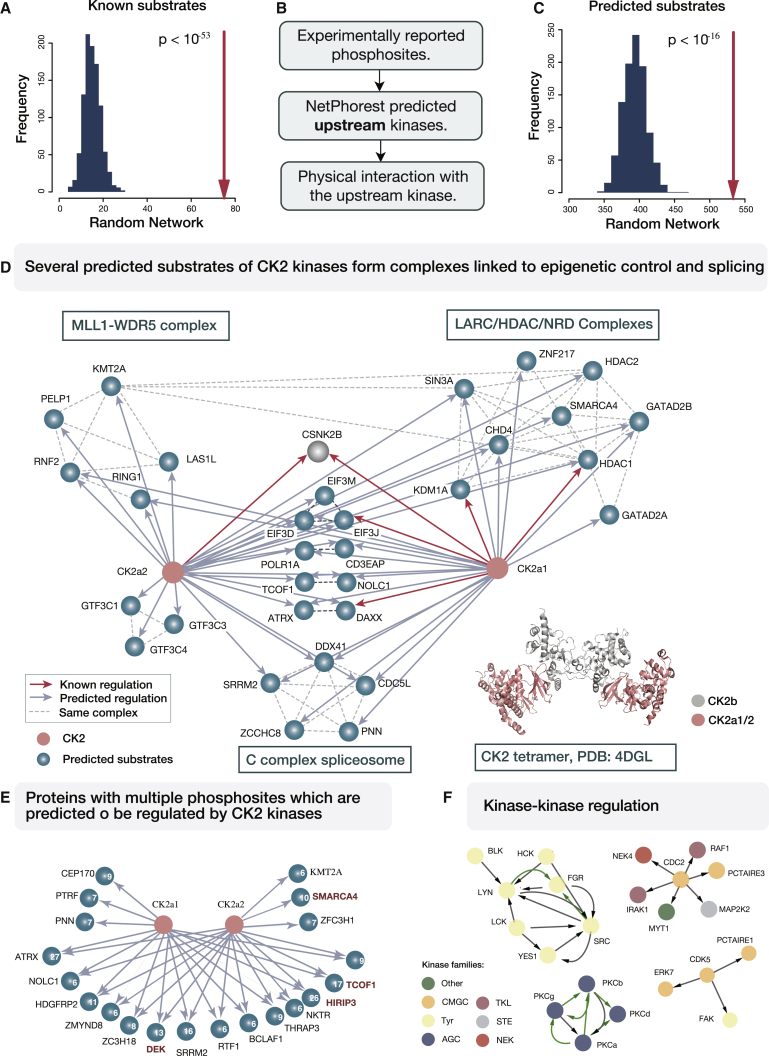
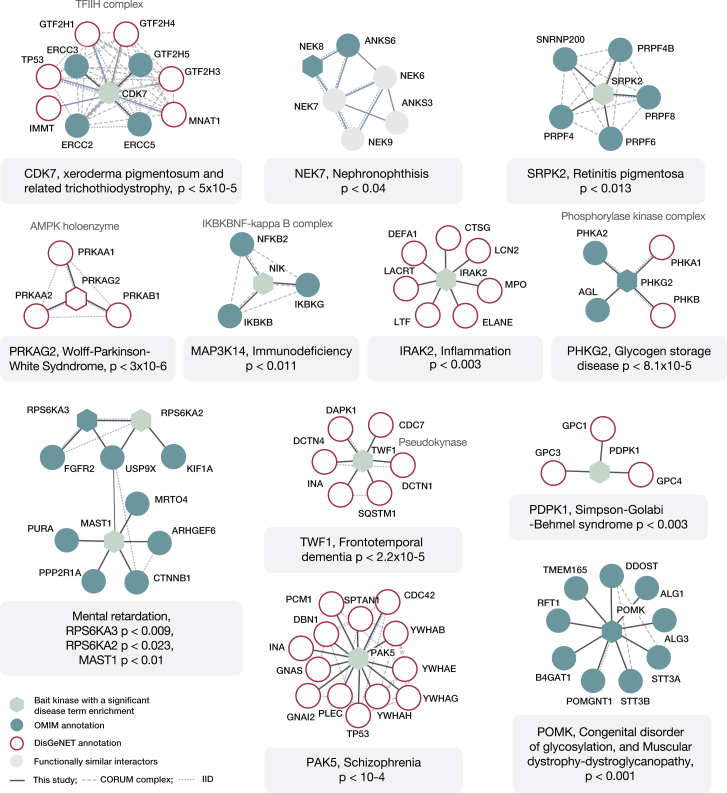
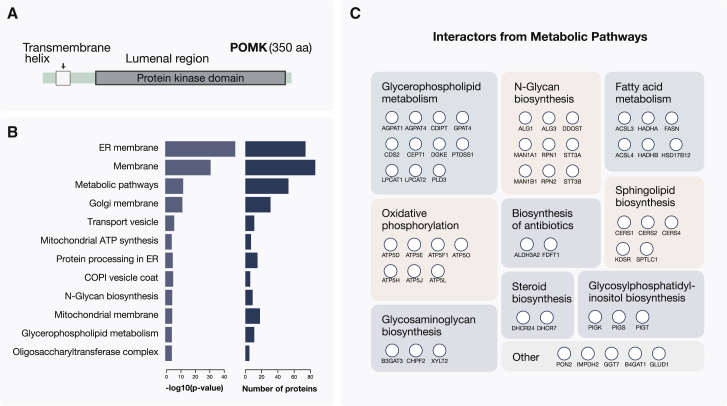
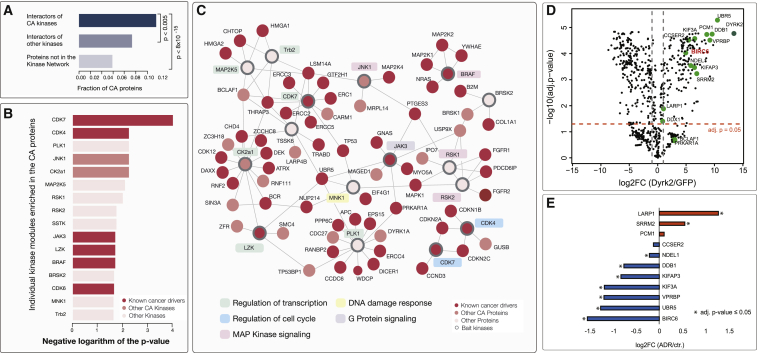
References
-
- Assrir N., Filhol O., Galisson F., Lipinski M. HIRIP3 is a nuclear phosphoprotein interacting with and phosphorylated by the serine-threonine kinase CK2. Biol. Chem. 2007;388:391–398. - PubMed
-
- Bensimon A., Aebersold R., Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–1639. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
