

Emergence of a High-Plasticity Cell State during Lung Cancer Evolution
- PMID: 32707077
- PMCID: PMC7745838
- DOI: 10.1016/j.ccell.2020.06.012
Emergence of a High-Plasticity Cell State during Lung Cancer Evolution
Abstract
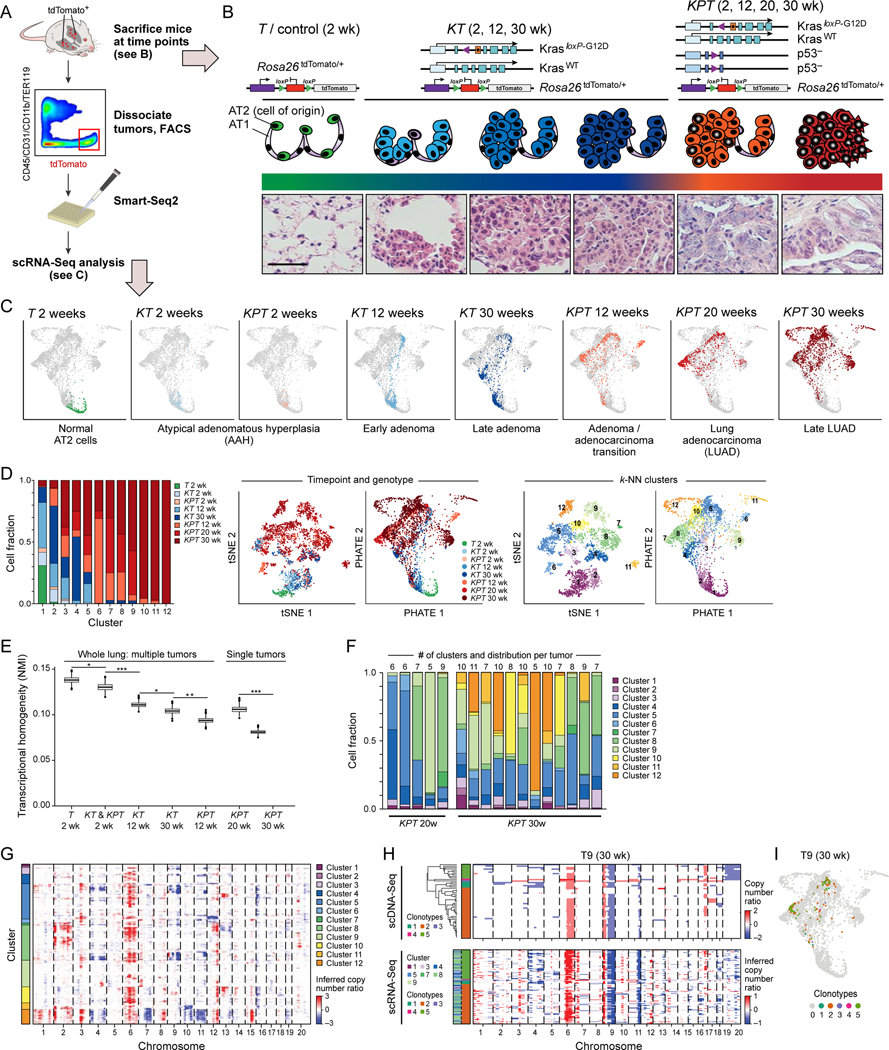
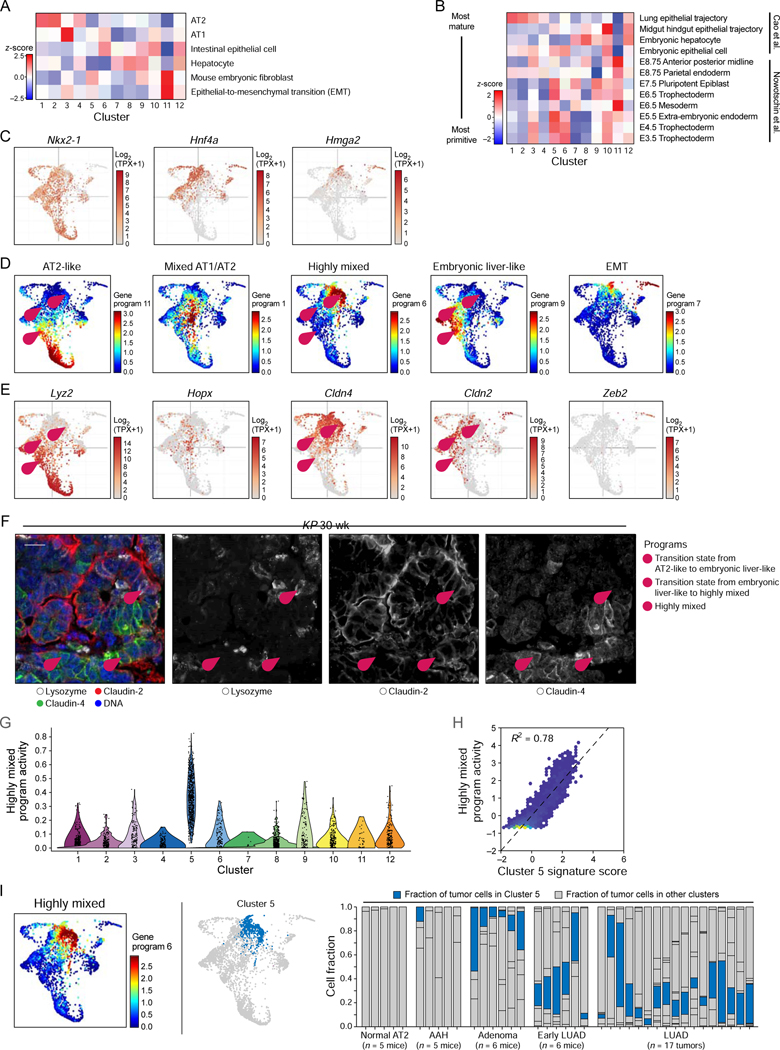
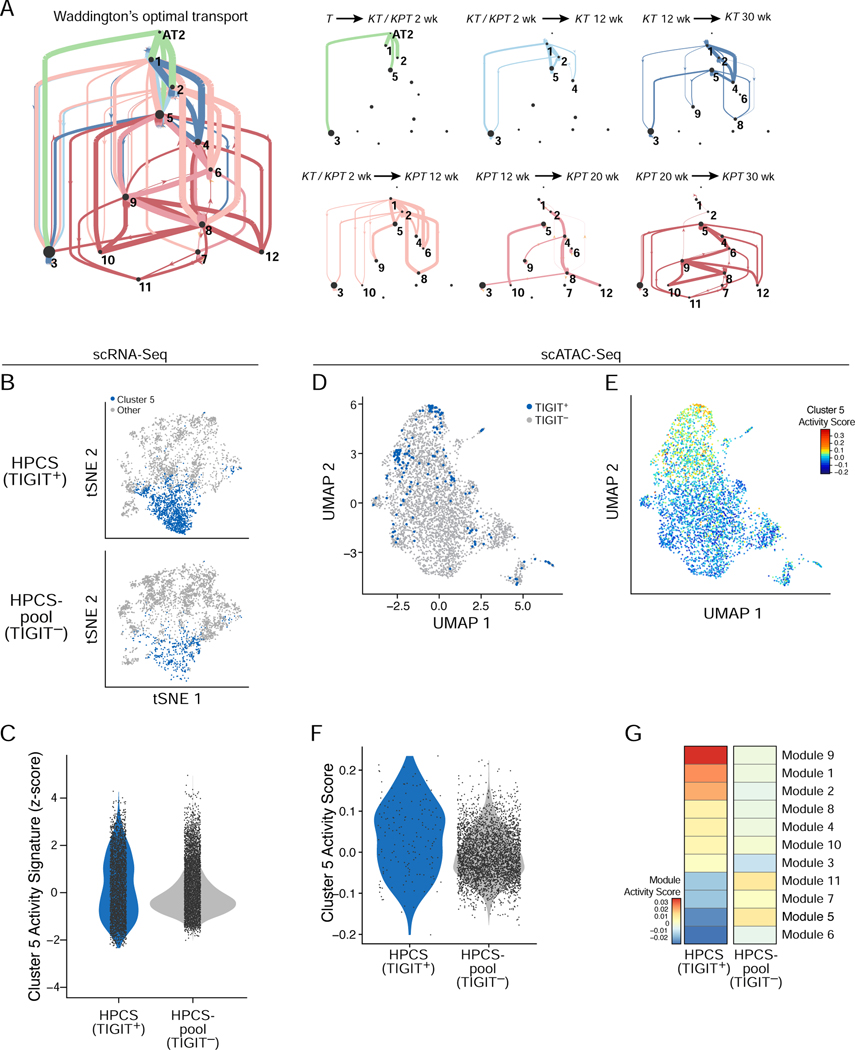
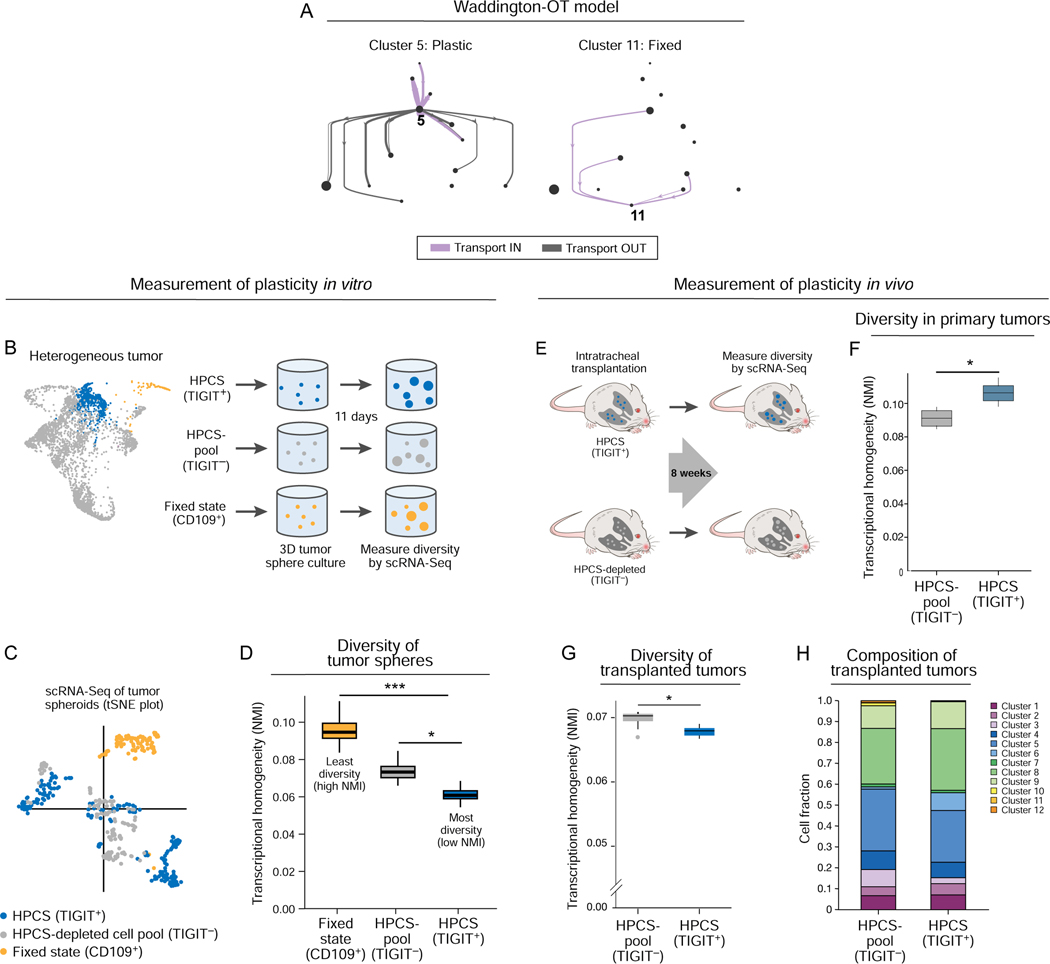
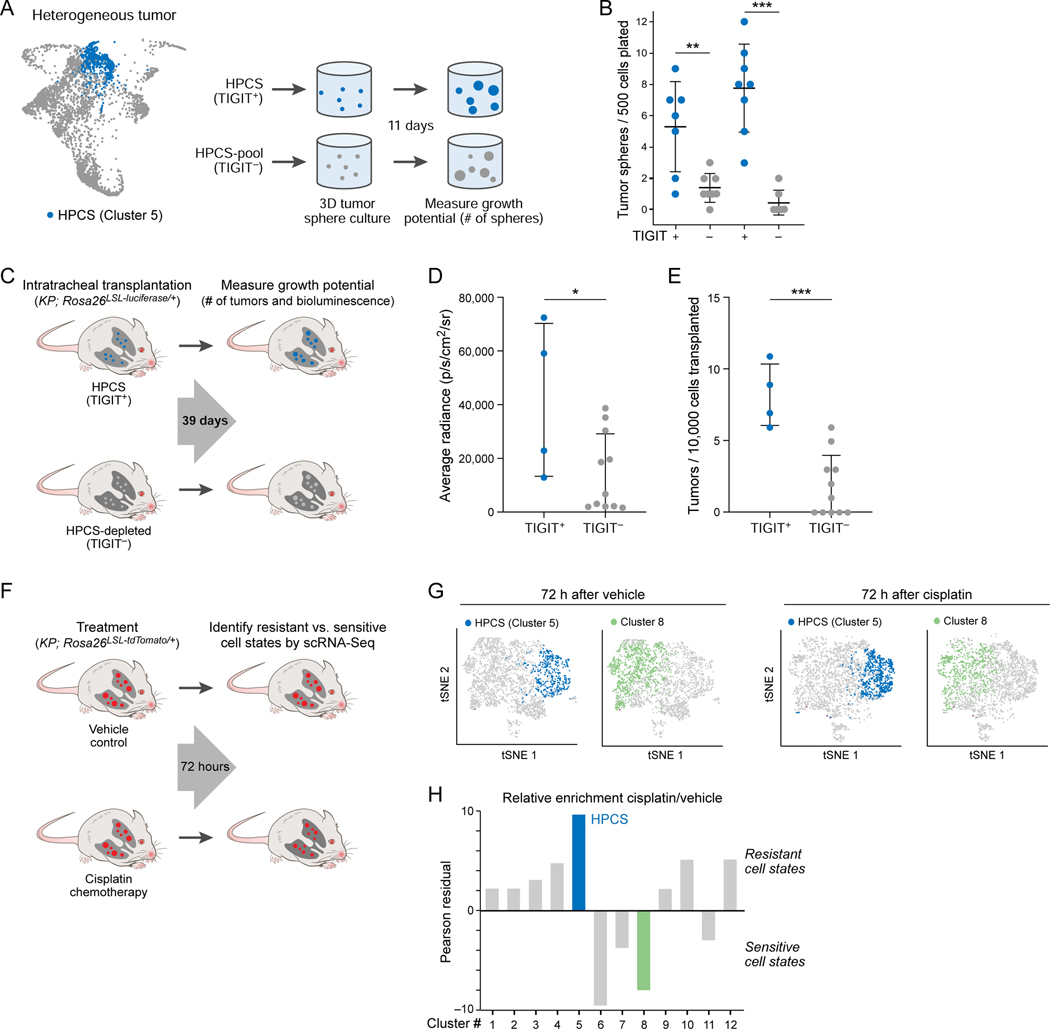
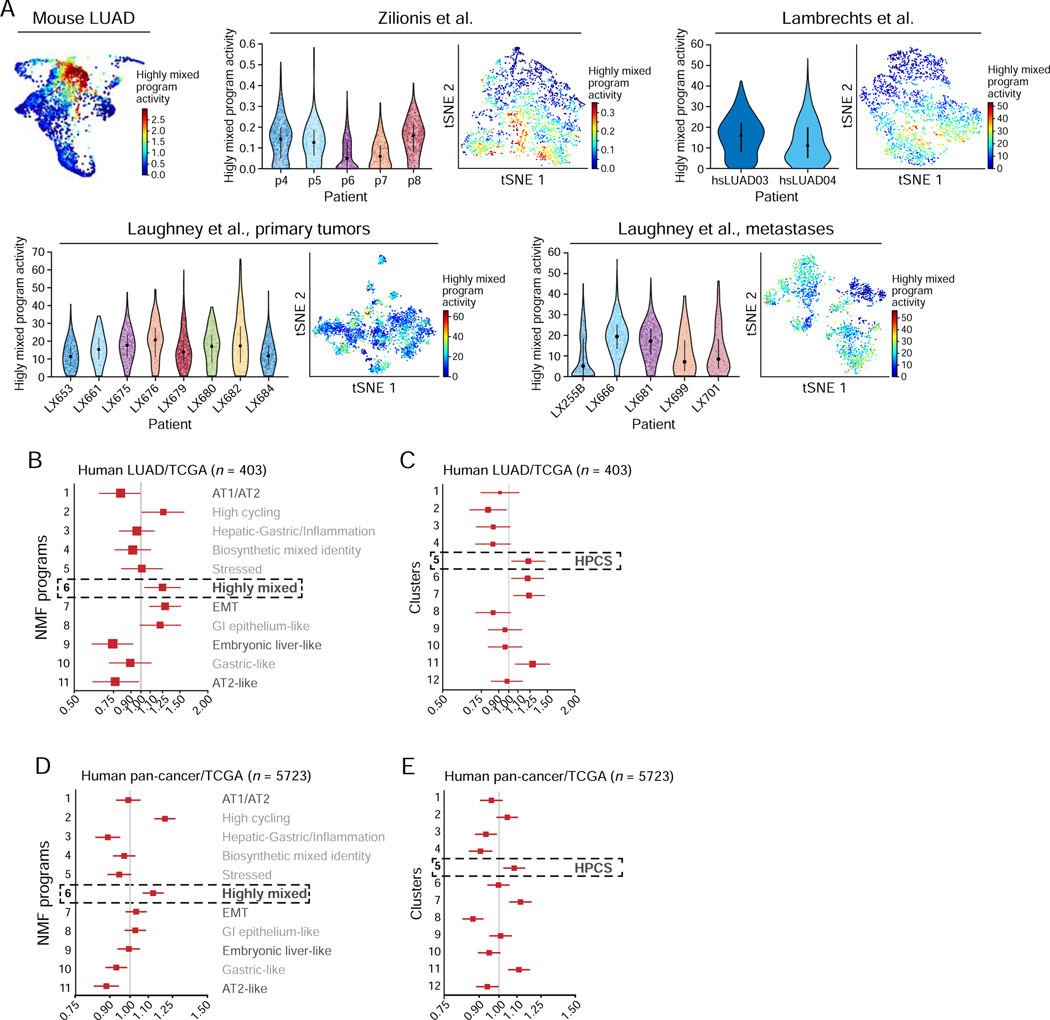
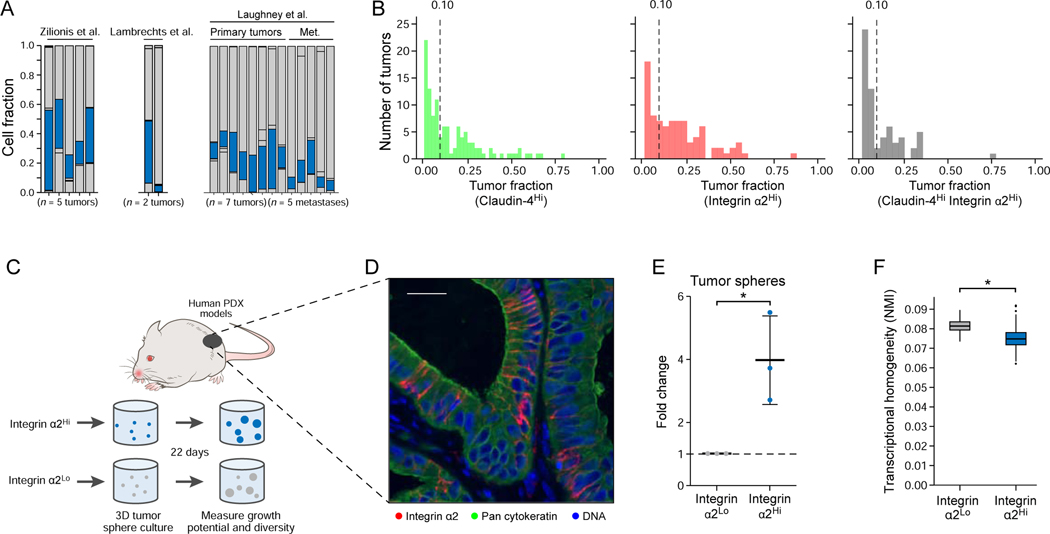
Tumor evolution from a single cell into a malignant, heterogeneous tissue remains poorly understood. Here, we profile single-cell transcriptomes of genetically engineered mouse lung tumors at seven stages, from pre-neoplastic hyperplasia to adenocarcinoma. The diversity of transcriptional states increases over time and is reproducible across tumors and mice. Cancer cells progressively adopt alternate lineage identities, computationally predicted to be mediated through a common transitional, high-plasticity cell state (HPCS). Accordingly, HPCS cells prospectively isolated from mouse tumors and human patient-derived xenografts display high capacity for differentiation and proliferation. The HPCS program is associated with poor survival across human cancers and demonstrates chemoresistance in mice. Our study reveals a central principle underpinning intra-tumoral heterogeneity and motivates therapeutic targeting of the HPCS.
Keywords: cell state transition; chromatin state; differentiation; drug resistance; lung cancer; plasticity; single-cell transcriptomics; tumor evolution; tumor heterogeneity; tumor progression.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. T.J. also received funding from Calico and currently receives funding from Johnson & Johnson, but this funding did not support the research described in this manuscript. A.R. is a co-founder and equity holder in Celsius Therapeutics and a SAB member for Thermo Fisher, Asimov, Neogene Therapeutics, and Syros Pharmaceuticals, and an equity holder of Immunitas Therapeutics. C.M.R. serves on the SAB of Bridge Medicines and Harpoon Therapeutics, and has consulted regarding oncology drug development with AbbVie, Amgen, Ascentage, Bicycle, Celgene, Daiichi Sankyo, Genentech, Ipsen, Loxo, Pharmamar, and Vavotek. None of the affiliations listed above represent a conflict of interest with the design or execution of this study or interpretation of data presented in this manuscript. Other authors have nothing to disclose.
Figures
Comment in
-
Studying Lineage Plasticity One Cell at a Time.Cancer Cell. 2020 Aug 10;38(2):150-152. doi: 10.1016/j.ccell.2020.07.001. Cancer Cell. 2020. PMID: 32781039
References
-
- Ambrogio C, Gomez-Lopez G, Falcone M, Vidal A, Nadal E, Crosetto N, Blasco RB, Fernandez-Marcos PJ, Sanchez-Cespedes M, Ren X, et al. (2016). Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med 22, 270–277. - PubMed
-
- Arozarena I, and Wellbrock C. (2019). Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer 19, 377–391. - PubMed
-
- Batlle E, and Clevers H. (2017). Cancer stem cells revisited. Nat Med 23, 1124–1134. - PubMed
-
- Bystrykh L, Weersing E, Dontje B, Sutton S, Pletcher MT, Wiltshire T, Su AI, Vellenga E, Wang J, Manly KF, et al. (2005). Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat Genet 37, 225–232. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
