

Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma
- PMID: 32707078
- PMCID: PMC7641015
- DOI: 10.1016/j.ccell.2020.06.006
Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma
Erratum in
-
Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma.Cancer Cell. 2025 May 12;43(5):981-984. doi: 10.1016/j.ccell.2025.02.025. Epub 2025 Mar 6. Cancer Cell. 2025. PMID: 40054468 No abstract available.
Abstract
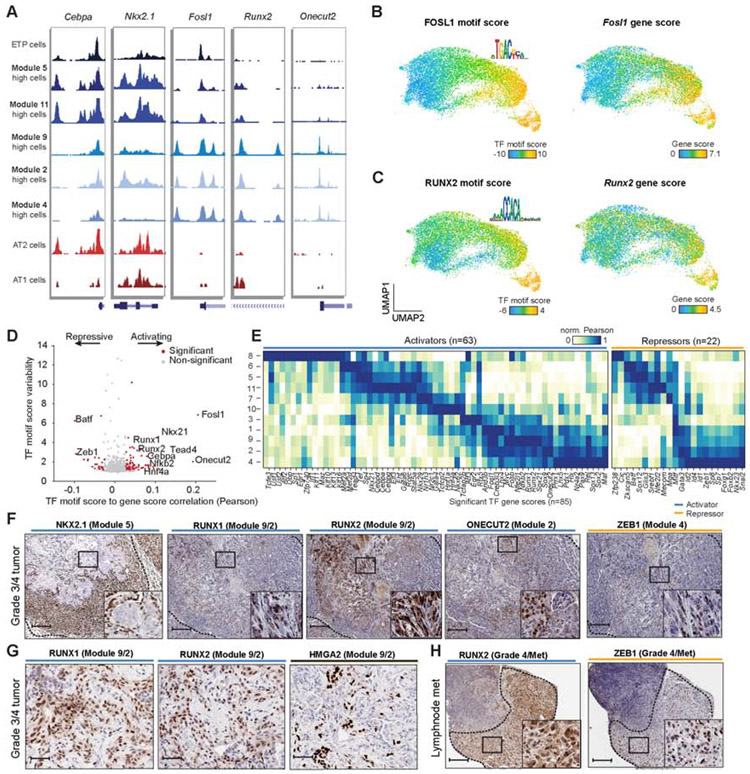
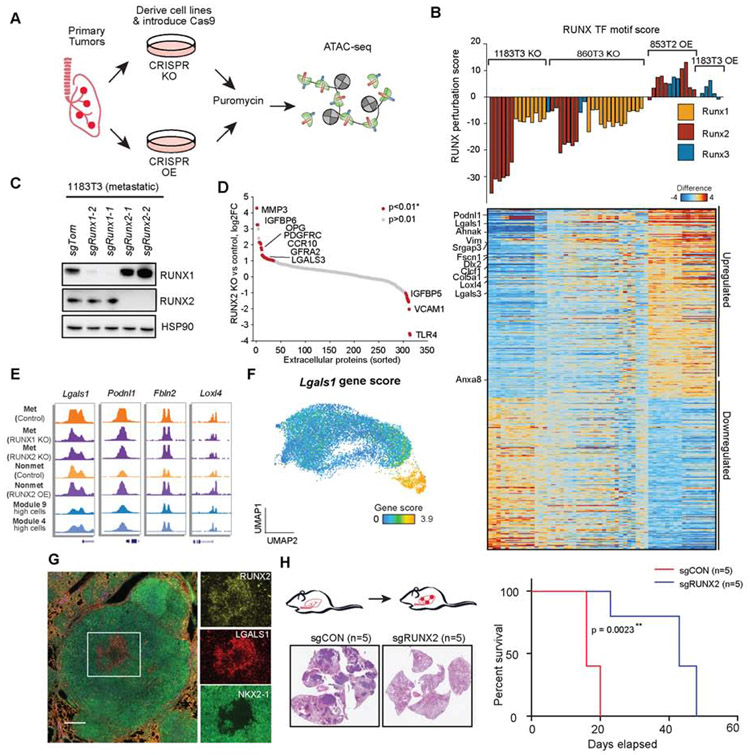
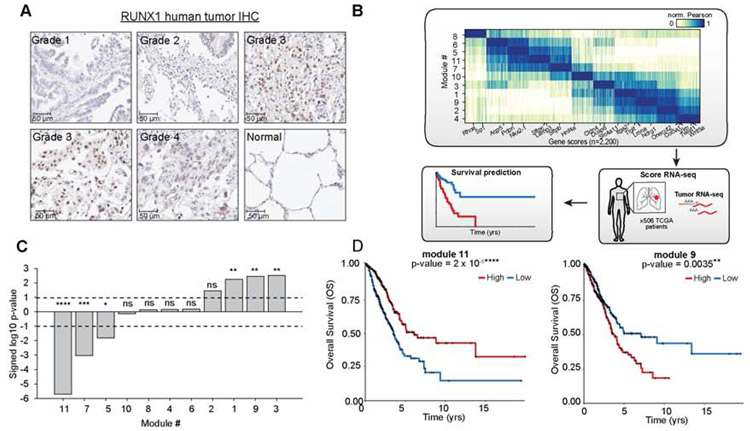
Regulatory networks that maintain functional, differentiated cell states are often dysregulated in tumor development. Here, we use single-cell epigenomics to profile chromatin state transitions in a mouse model of lung adenocarcinoma (LUAD). We identify an epigenomic continuum representing loss of cellular identity and progression toward a metastatic state. We define co-accessible regulatory programs and infer key activating and repressive chromatin regulators of these cell states. Among these co-accessibility programs, we identify a pre-metastatic transition, characterized by activation of RUNX transcription factors, which mediates extracellular matrix remodeling to promote metastasis and is predictive of survival across human LUAD patients. Together, these results demonstrate the power of single-cell epigenomics to identify regulatory programs to uncover mechanisms and key biomarkers of tumor progression.
Keywords: cancer; epigenomics; epithelial-to-mesenchymal transition; metastasis; non-small cell lung cancer; single cell.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific. He is also a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. T.J. laboratory currently also receives funding from the Johnson & Johnson Lung Cancer Initiative and The Lustgarten Foundation for Pancreatic Cancer Research, but this funding did not support the research described in this manuscript. J.D.B. holds patents related to ATAC-seq and single-cell ATAC-seq and serves on the Scientific Advisory Board of CAMP4 Therapeutics and seqWell. A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and an SAB member of Thermo Fisher Scientific, Syros Pharmaceuticals, Asimov, and Neogene Therapeutics.
Figures
Comment in
-
Studying Lineage Plasticity One Cell at a Time.Cancer Cell. 2020 Aug 10;38(2):150-152. doi: 10.1016/j.ccell.2020.07.001. Cancer Cell. 2020. PMID: 32781039
References
-
- Bai X, Meng L, Sun H, Li Z, Zhang X, and Hua S (2017). MicroRNA-196b Inhibits Cell Growth and Metastasis of Lung Cancer Cells by Targeting Runx2. Cell. Physiol. Biochem 43, 757–767. - PubMed
-
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
