

Genomic Islands in Mycoplasmas
- PMID: 32707922
- PMCID: PMC7466169
- DOI: 10.3390/genes11080836
Genomic Islands in Mycoplasmas
Abstract
Bacteria of the Mycoplasma genus are characterized by the lack of a cell-wall, the use of UGA as tryptophan codon instead of a universal stop, and their simplified metabolic pathways. Most of these features are due to the small-size and limited-content of their genomes (580-1840 Kbp; 482-2050 CDS). Yet, the Mycoplasma genus encompasses over 200 species living in close contact with a wide range of animal hosts and man. These include pathogens, pathobionts, or commensals that have retained the full capacity to synthesize DNA, RNA, and all proteins required to sustain a parasitic life-style, with most being able to grow under laboratory conditions without host cells. Over the last 10 years, comparative genome analyses of multiple species and strains unveiled some of the dynamics of mycoplasma genomes. This review summarizes our current knowledge of genomic islands (GIs) found in mycoplasmas, with a focus on pathogenicity islands, integrative and conjugative elements (ICEs), and prophages. Here, we discuss how GIs contribute to the dynamics of mycoplasma genomes and how they participate in the evolution of these minimal organisms.
Keywords: Mollicutes; evolution; genome; genomic island; horizontal gene transfer; mobile elements; mycoplasmas; phages.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
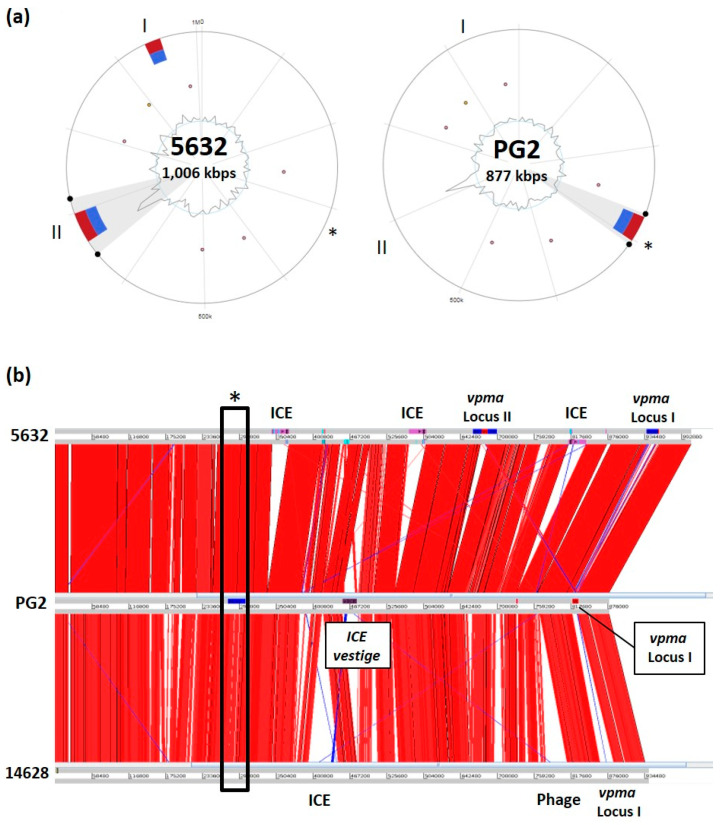
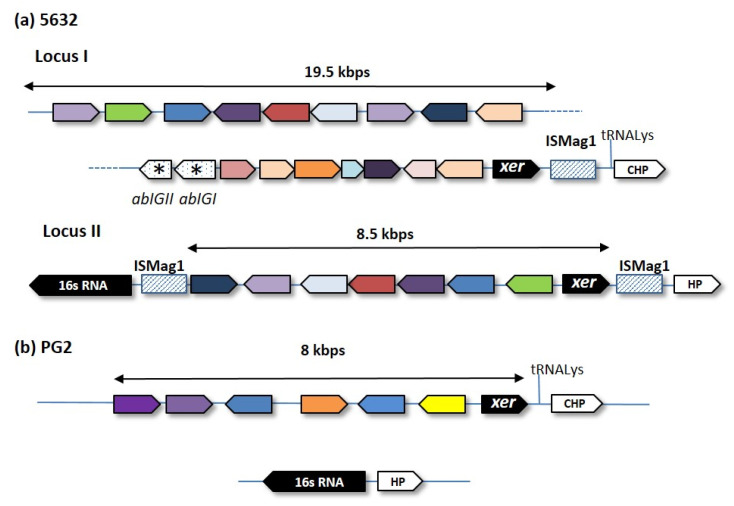
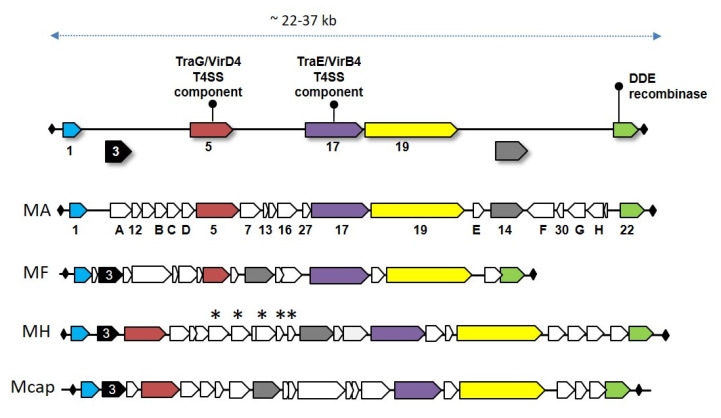

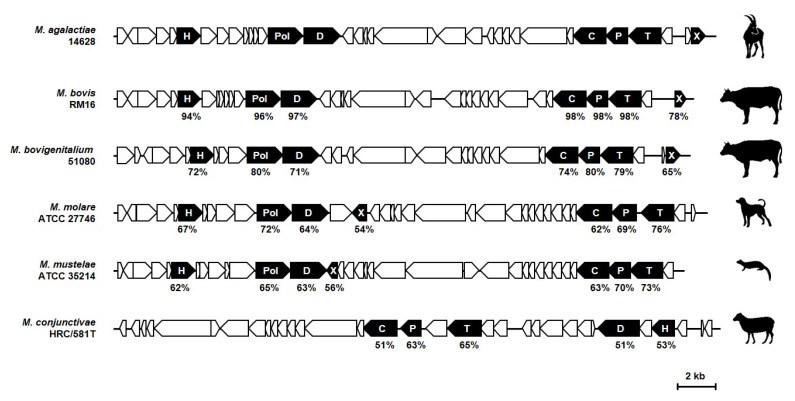
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
