

HDX-MS: An Analytical Tool to Capture Protein Motion in Action
- PMID: 32709043
- PMCID: PMC7399943
- DOI: 10.3390/biomedicines8070224
HDX-MS: An Analytical Tool to Capture Protein Motion in Action
Abstract
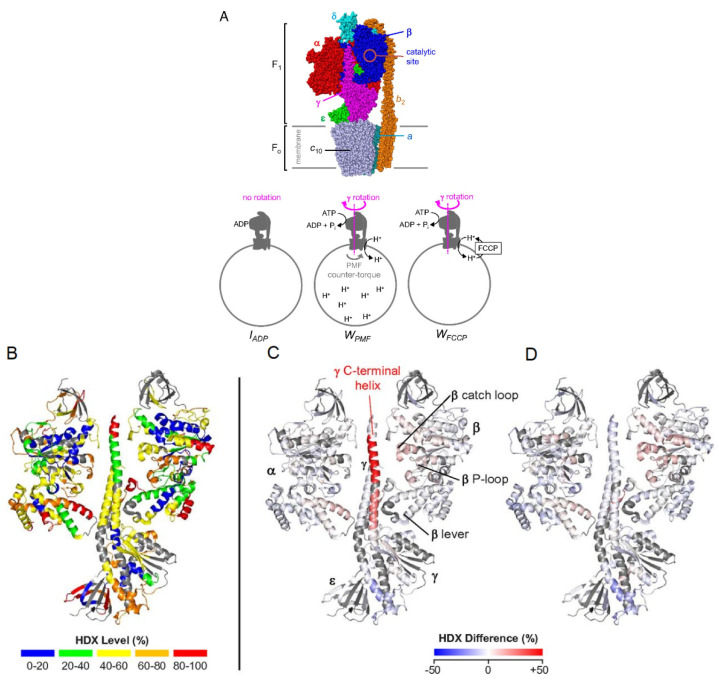
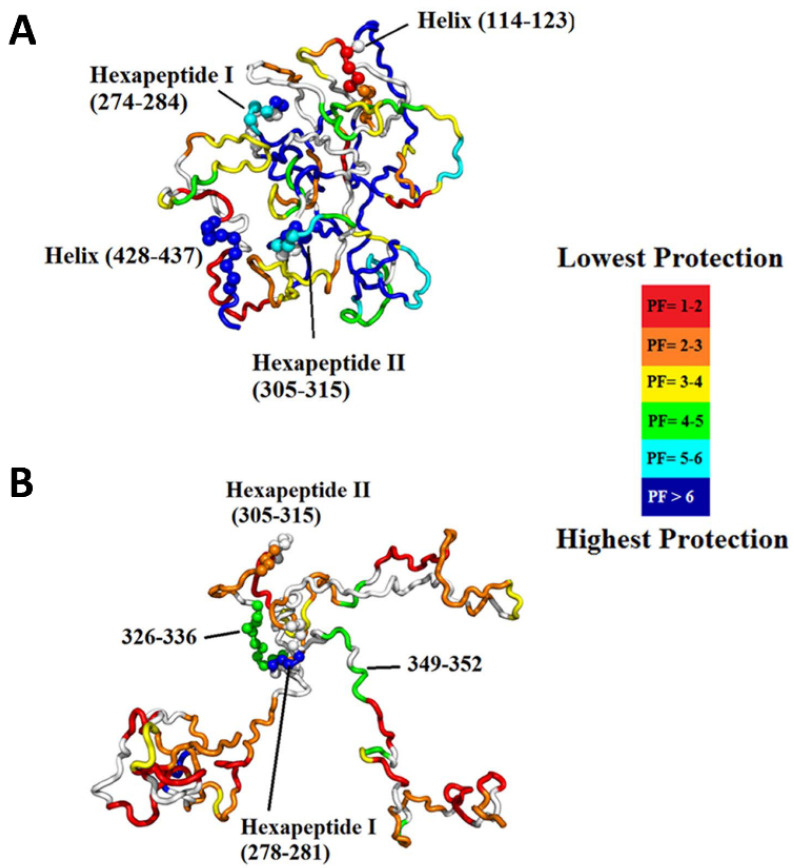
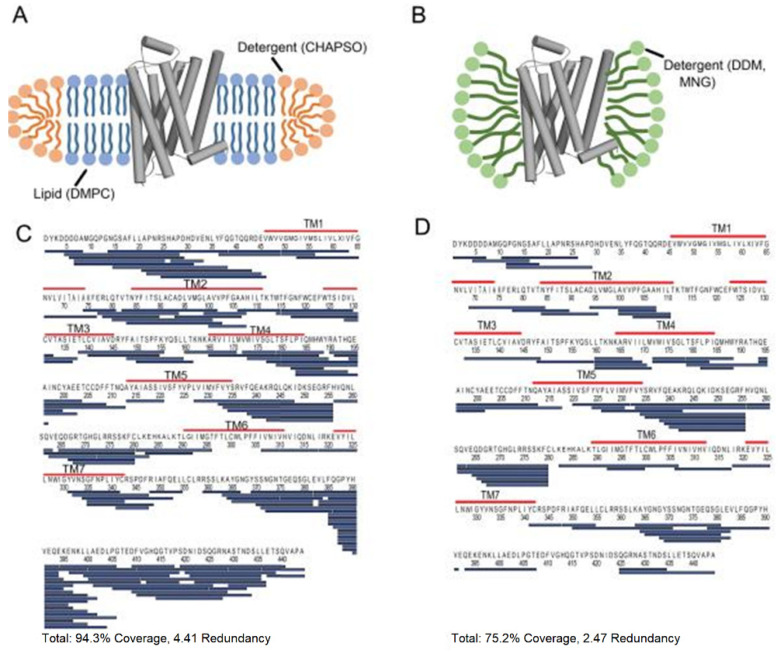
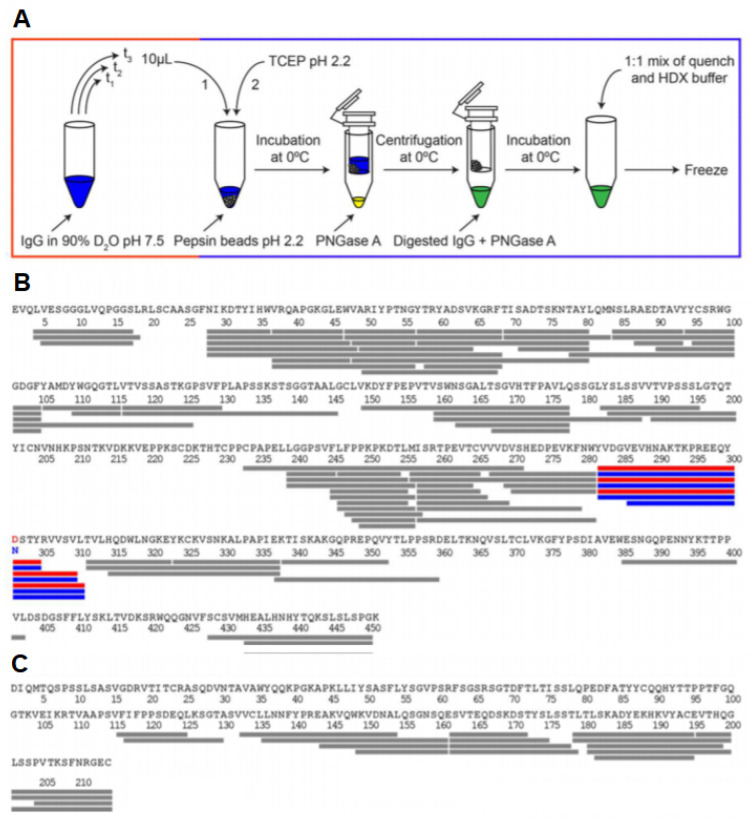
Virtually all protein functions in the cell, including pathogenic processes, require coordinated motion of atoms or domains, i.e., conformational dynamics. Understanding protein dynamics is therefore critical both for drug development and to learn about the underlying molecular causes of many diseases. Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) provides valuable information about protein dynamics, which is highly complementary to the static picture provided by conventional high-resolution structural tools (i.e., X-ray crystallography and structural NMR). The amount of protein required to carry out HDX-MS experiments is a fraction of the amount required by alternative biophysical techniques, which are also usually lower resolution. Use of HDX-MS is growing quickly both in industry and academia, and it has been successfully used in numerous drug and vaccine development efforts, with important roles in understanding allosteric effects and mapping binding sites.
Keywords: HDX-MS; hydrogen–deuterium exchange mass spectrometry; protein dynamics; protein folding and misfolding.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
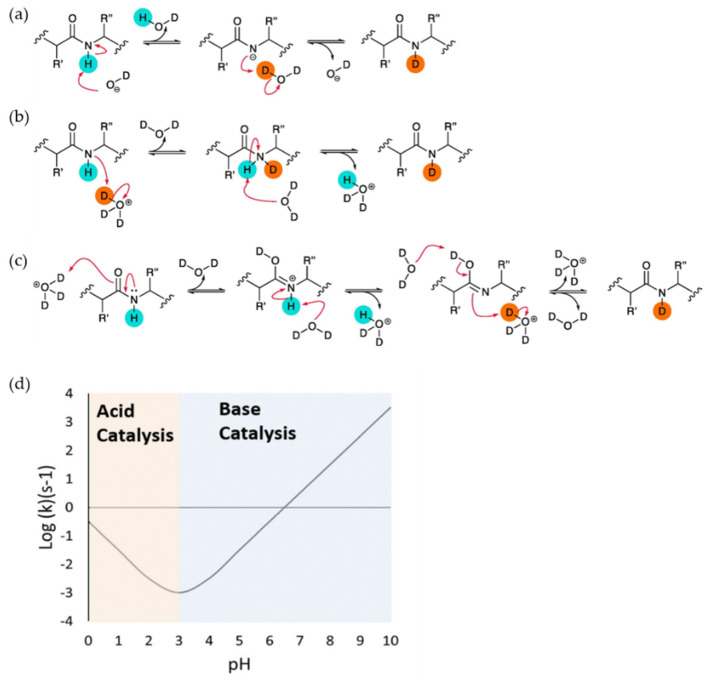
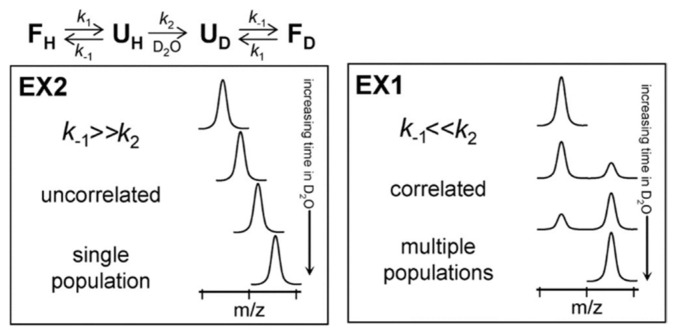
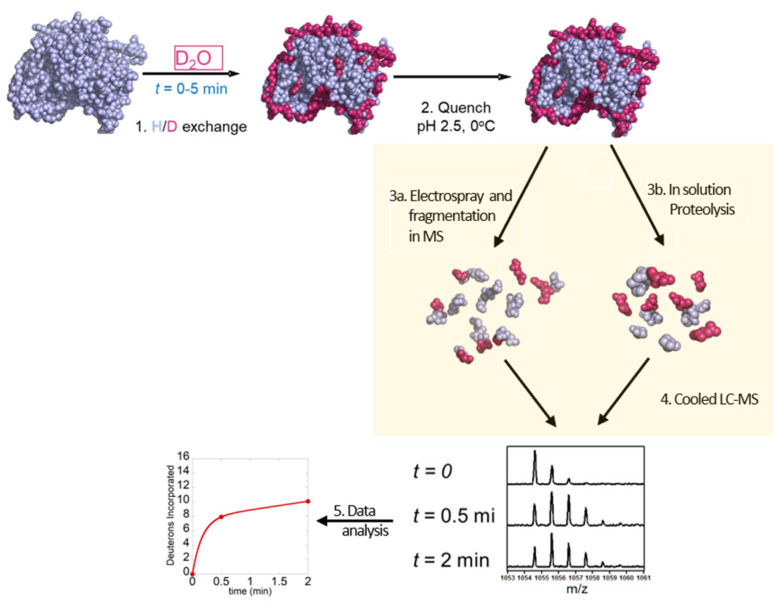
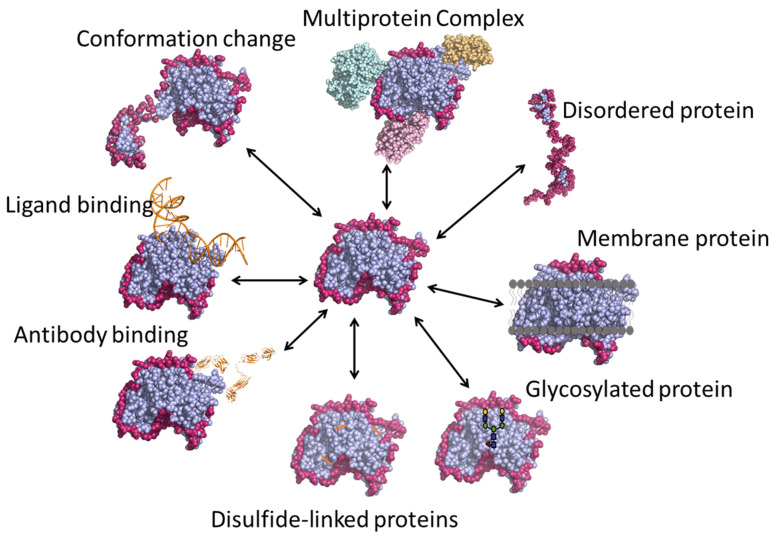
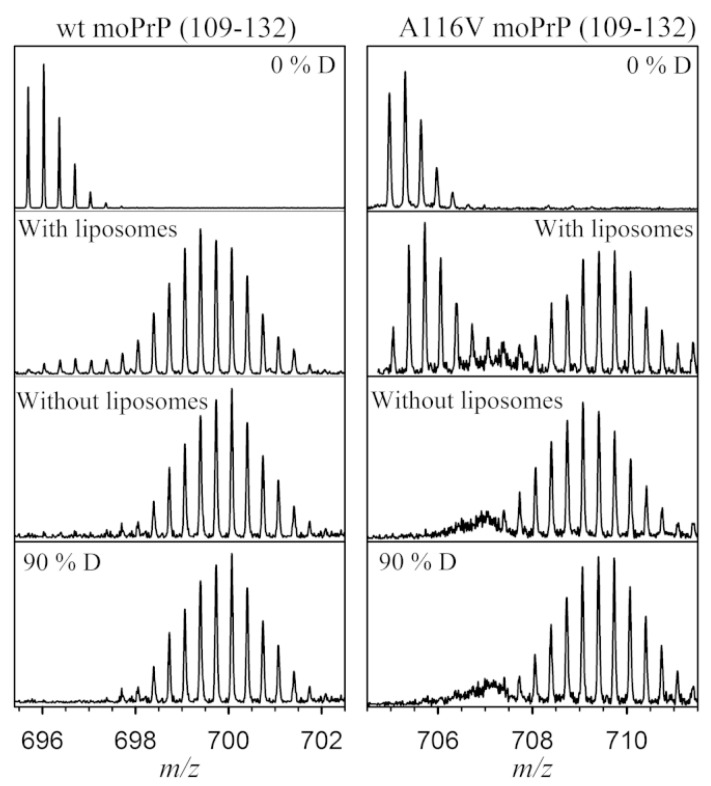
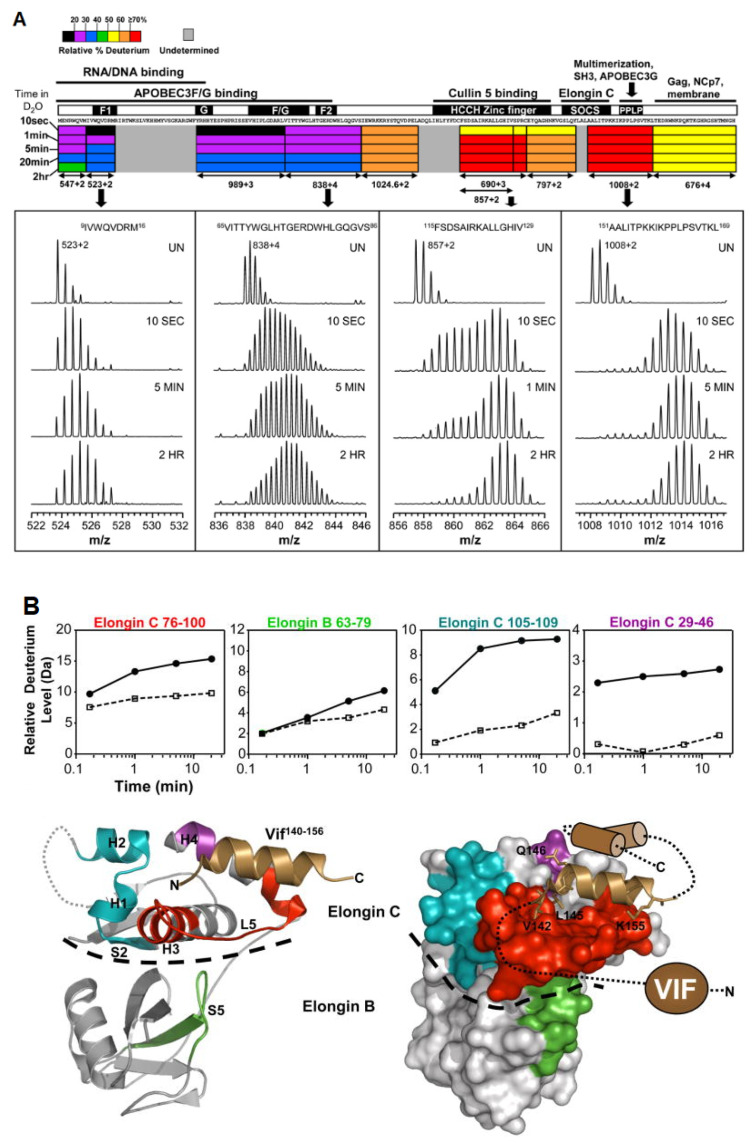
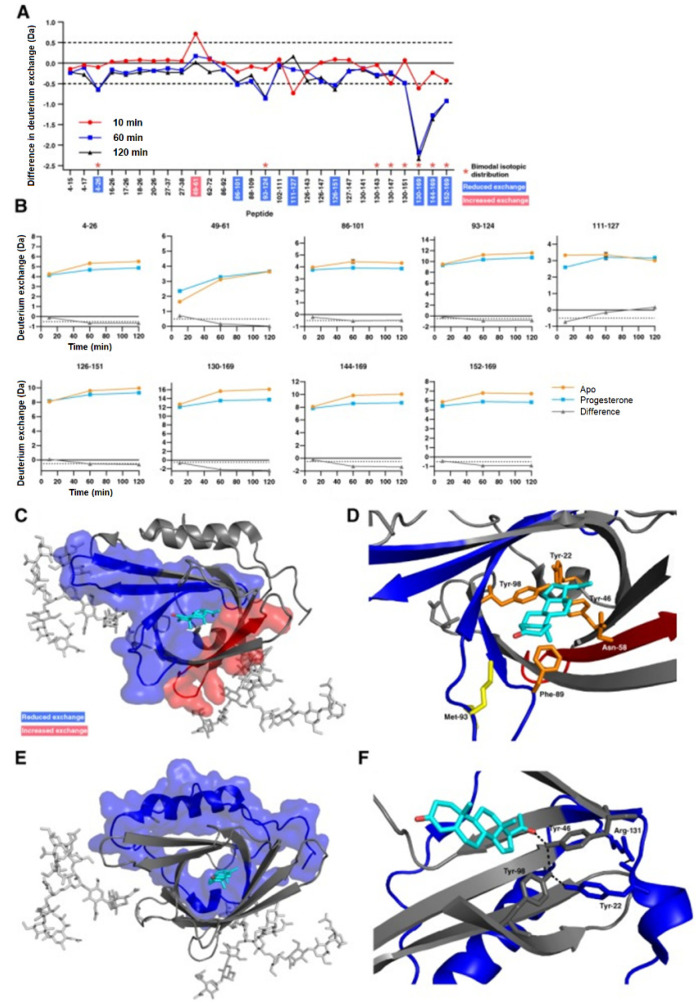
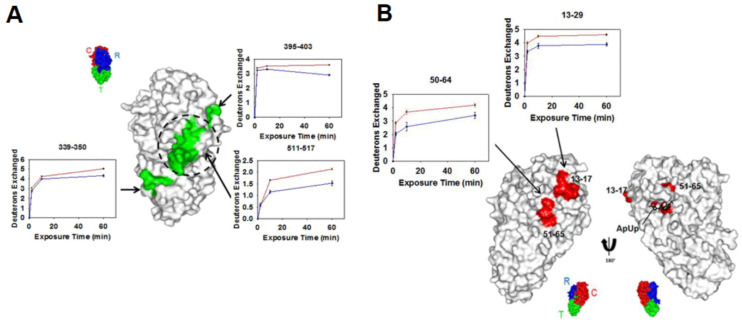
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
