

Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer
- PMID: 32709844
- PMCID: PMC7381637
- DOI: 10.1038/s41467-020-17492-y
Single-cell transcriptomic analysis in a mouse model deciphers cell transition states in the multistep development of esophageal cancer
Abstract
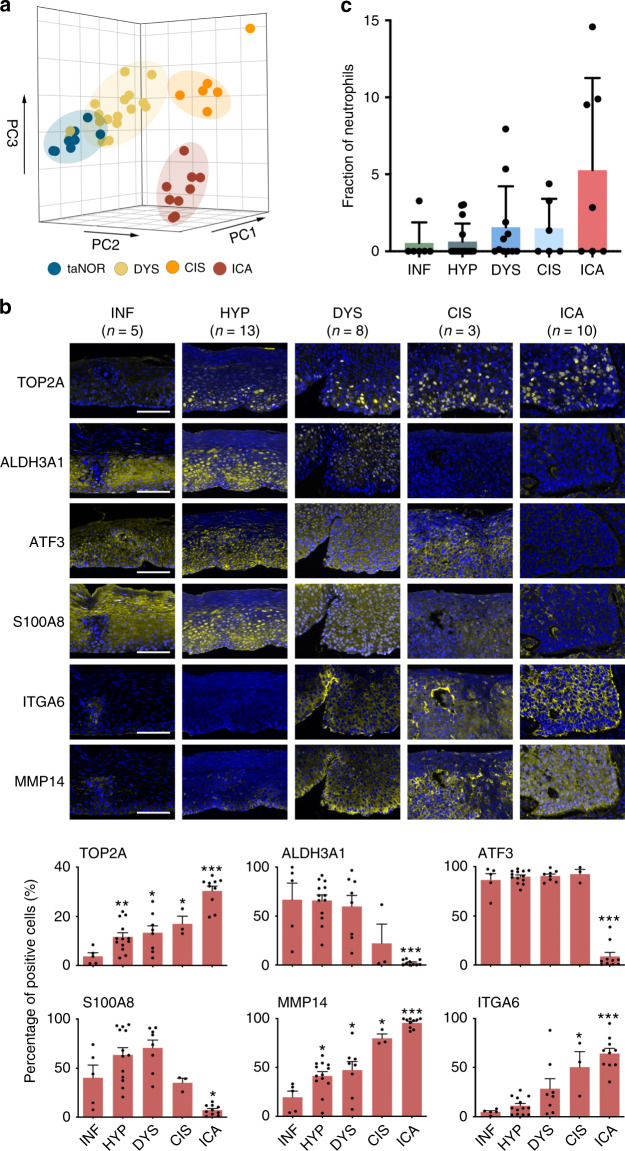
Esophageal squamous cell carcinoma (ESCC) is prevalent in some geographical regions of the world. ESCC development presents a multistep pathogenic process from inflammation to invasive cancer; however, what is critical in these processes and how they evolve is largely unknown, obstructing early diagnosis and effective treatment. Here, we create a mouse model mimicking human ESCC development and construct a single-cell ESCC developmental atlas. We identify a set of key transitional signatures associated with oncogenic evolution of epithelial cells and depict the landmark dynamic tumorigenic trajectories. An early downregulation of CD8+ response against the initial tissue damage accompanied by the transition of immune response from type 1 to type 3 results in accumulation and activation of macrophages and neutrophils, which may create a chronic inflammatory environment that promotes carcinogen-transformed epithelial cell survival and proliferation. These findings shed light on how ESCC is initiated and developed.
Conflict of interest statement
The authors declare no competing interests.
Figures






References
-
- Gao Y, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014;46:1097–1102. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
