

DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation
- PMID: 32709850
- PMCID: PMC7381644
- DOI: 10.1038/s41467-020-17531-8
DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation
Abstract
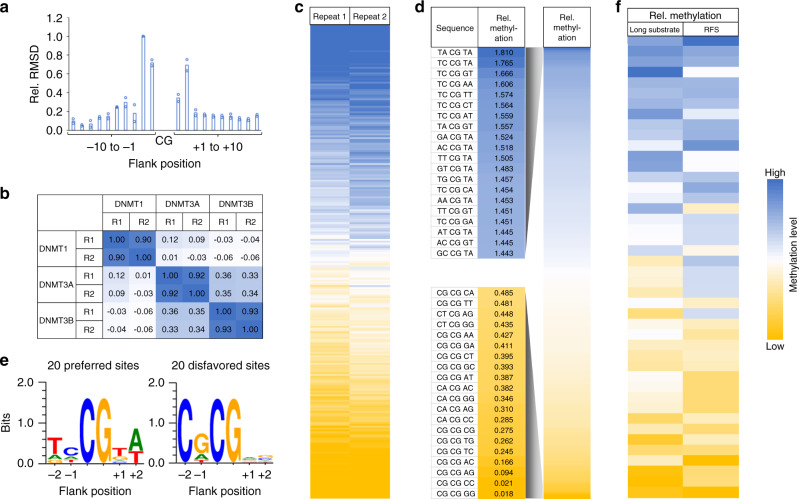
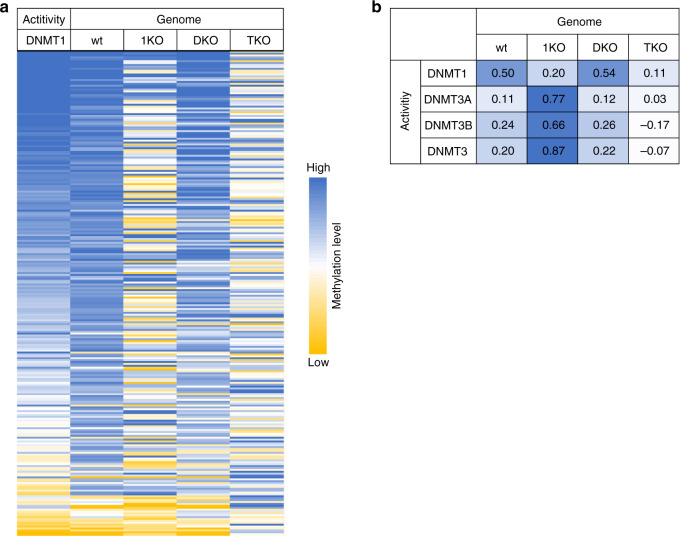
DNA methylation maintenance by DNMT1 is an essential process in mammals but molecular mechanisms connecting DNA methylation patterns and enzyme activity remain elusive. Here, we systematically analyzed the specificity of DNMT1, revealing a pronounced influence of the DNA sequences flanking the target CpG site on DNMT1 activity. We determined DNMT1 structures in complex with preferred DNA substrates revealing that DNMT1 employs flanking sequence-dependent base flipping mechanisms, with large structural rearrangements of the DNA correlating with low catalytic activity. Moreover, flanking sequences influence the conformational dynamics of the active site and cofactor binding pocket. Importantly, we show that the flanking sequence preferences of DNMT1 highly correlate with genomic methylation in human and mouse cells, and 5-azacytidine triggered DNA demethylation is more pronounced at CpG sites with flanks disfavored by DNMT1. Overall, our findings uncover the intricate interplay between CpG-flanking sequence, DNMT1-mediated base flipping and the dynamic landscape of DNA methylation.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. - PubMed
-
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. - PubMed
-
- Jabbari K, Bernardi G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene. 2004;333:143–149. - PubMed
-
- Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. - PubMed
