

Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models
- PMID: 32709856
- PMCID: PMC7381609
- DOI: 10.1038/s41467-020-17127-2
Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models
Abstract
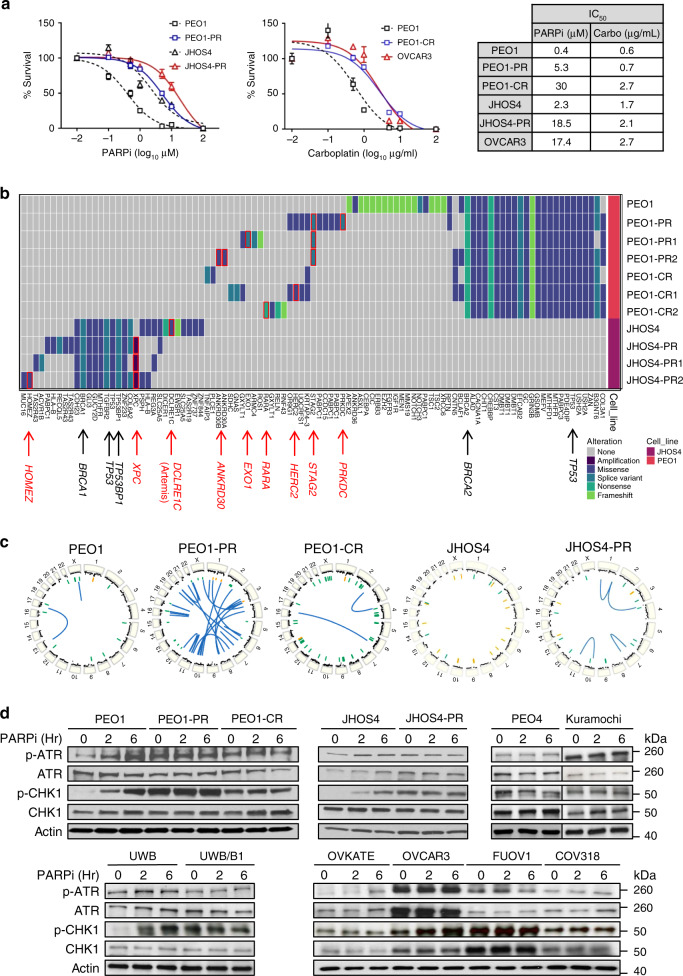
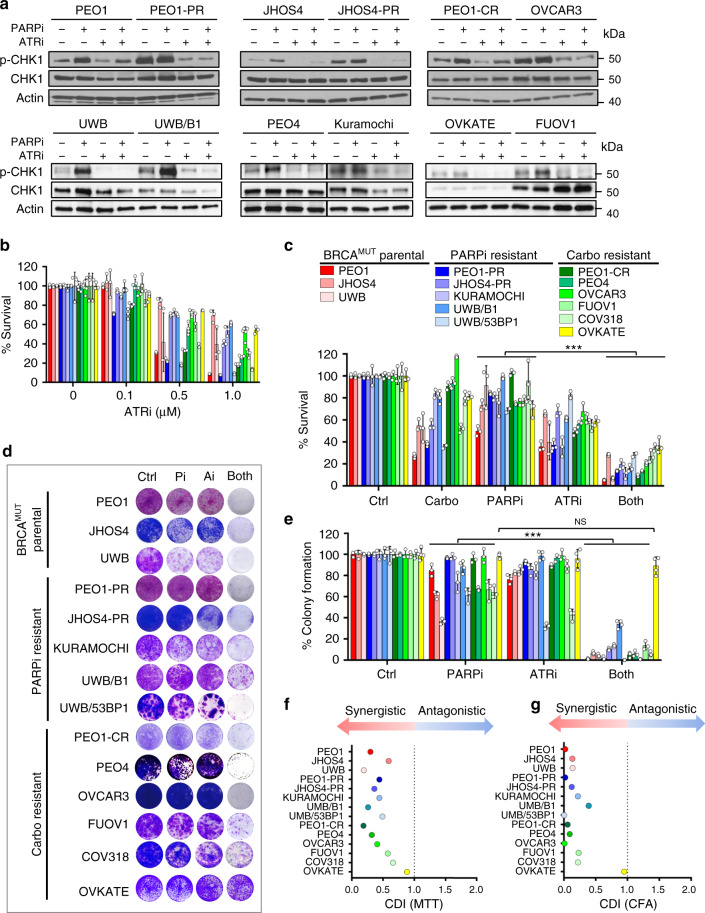
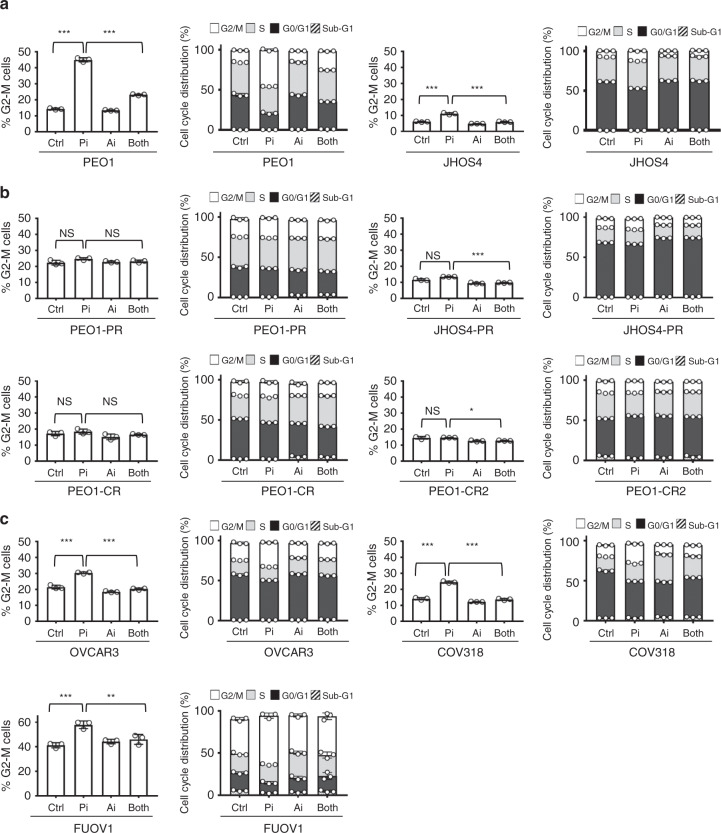
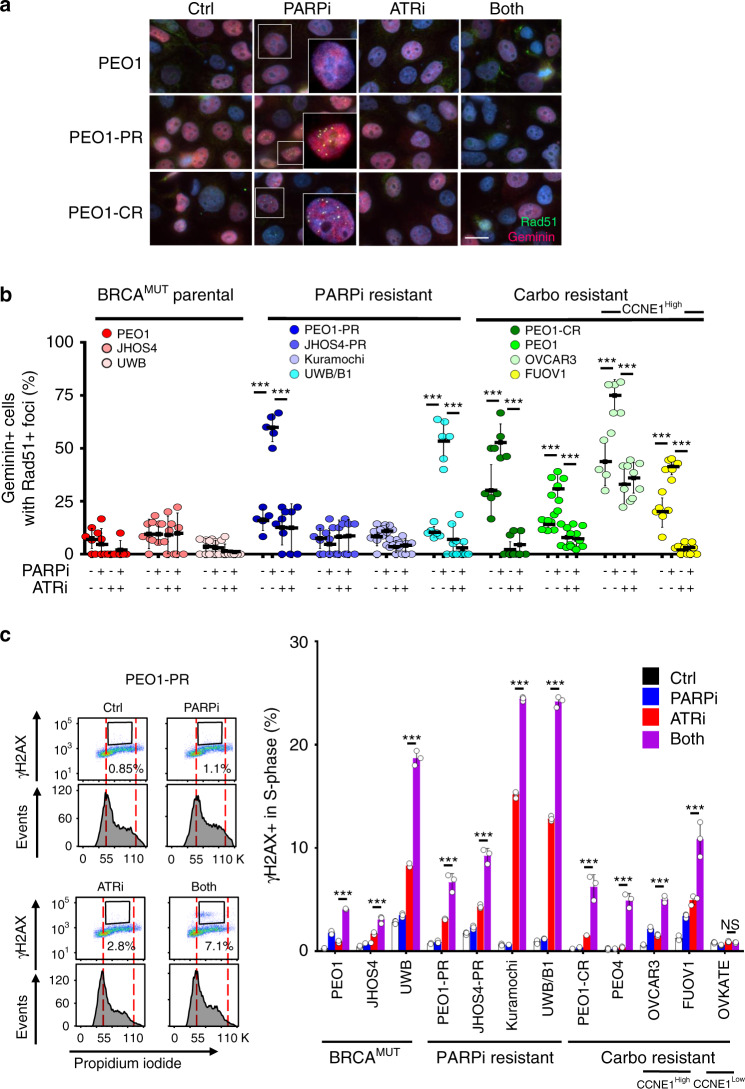
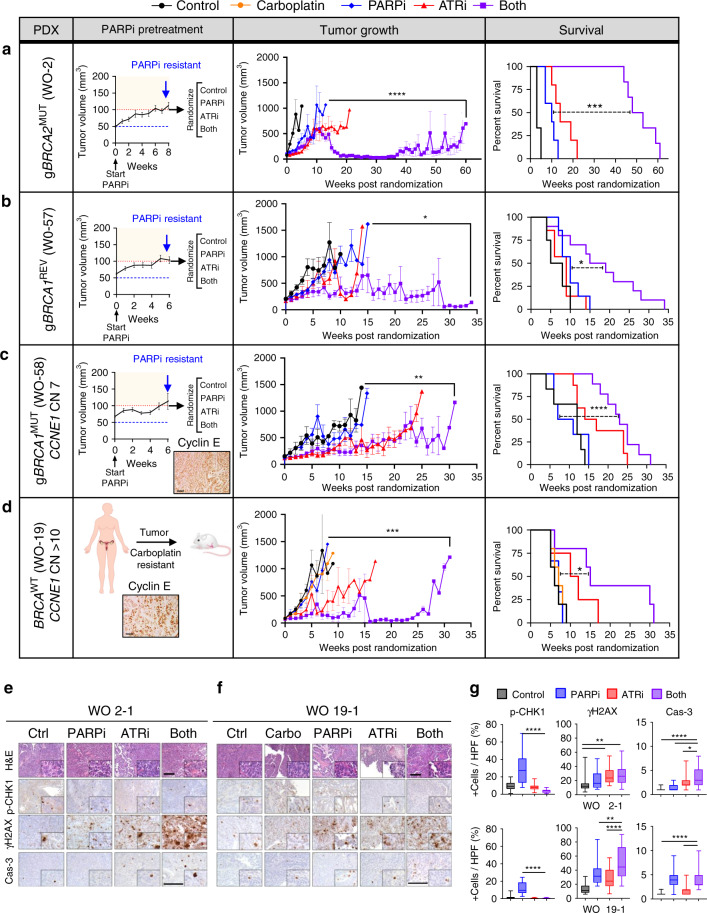
Ovarian cancer (OVCA) inevitably acquires resistance to platinum chemotherapy and PARP inhibitors (PARPi). We show that acquisition of PARPi-resistance is accompanied by increased ATR-CHK1 activity and sensitivity to ATR inhibition (ATRi). However, PARPi-resistant cells are remarkably more sensitive to ATRi when combined with PARPi (PARPi-ATRi). Sensitivity to PARPi-ATRi in diverse PARPi and platinum-resistant models, including BRCA1/2 reversion and CCNE1-amplified models, correlate with synergistic increases in replication fork stalling, double-strand breaks, and apoptosis. Surprisingly, BRCA reversion mutations and an ability to form RAD51 foci are frequently not observed in models of acquired PARPi-resistance, suggesting the existence of alternative resistance mechanisms. However, regardless of the mechanisms of resistance, complete and durable therapeutic responses to PARPi-ATRi that significantly increase survival are observed in clinically relevant platinum and acquired PARPi-resistant patient-derived xenografts (PDXs) models. These findings indicate that PARPi-ATRi is a highly promising strategy for OVCAs that acquire resistance to PARPi and platinum.
Conflict of interest statement
The following authors declare competing interests. F.S. serves on the scientific advisory board for AstraZeneca (unpaid). E.J.B. serves on the scientific advisory board of Atrin Pharmaceuticals and has been an advisor for Sierra Oncology. RAG has consulted for Agios Pharmaceuticals and MoMa Therapeutics. V.E.V. is a founder of Personal Genome Diagnostics, a member of its Scientific Advisory Board and Board of Directors, and owns Personal Genome Diagnostics stock, which are subject to certain restrictions under university policy. V.E.V. is an advisor to Takeda Pharmaceuticals. Within the last 5 years, V.E.V. has been an advisor to Daiichi Sankyo, Janssen Diagnostics, and Ignyta. R.B.S. is a founder of and holds equity in Delfi Diagnostics. He also serves as the Head of Data Sciences at Delfi Diagnostics. The terms for these arrangements for V.E.V. and R.B.S. has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. G.B.M. receives support or acts as a consultant for AstraZeneca, ImmunoMET, Ionis, Nanostring, PDX Pharmaceuticals, Signalchem Lifesciences, Symphogen, and Tarveda and has transferred technology to Myriad and Nanostring. The remaining authors declare no competing interests.
Figures

References
-
- Salani R, et al. Posttreatment surveillance and diagnosis of recurrence in women with gynecologic malignancies: Society of Gynecologic Oncologists recommendations. Am. J. Obstet. Gynecol. 2011;204:466–478. - PubMed
-
- Zivanovic O, Aldini A, Carlson JW, Chi DS. Advanced cytoreductive surgery: American perspective. Gynecologic Oncol. 2009;114:S3–S9. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: Cancer J. Clin. 2018;68:7–30. - PubMed
-
- Kristeleit R, et al. A Phase I-II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin. Cancer Res. 2017;23:4095–4106. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
