

Tutorial: a guide to performing polygenic risk score analyses
- PMID: 32709988
- PMCID: PMC7612115
- DOI: 10.1038/s41596-020-0353-1
Tutorial: a guide to performing polygenic risk score analyses
Abstract
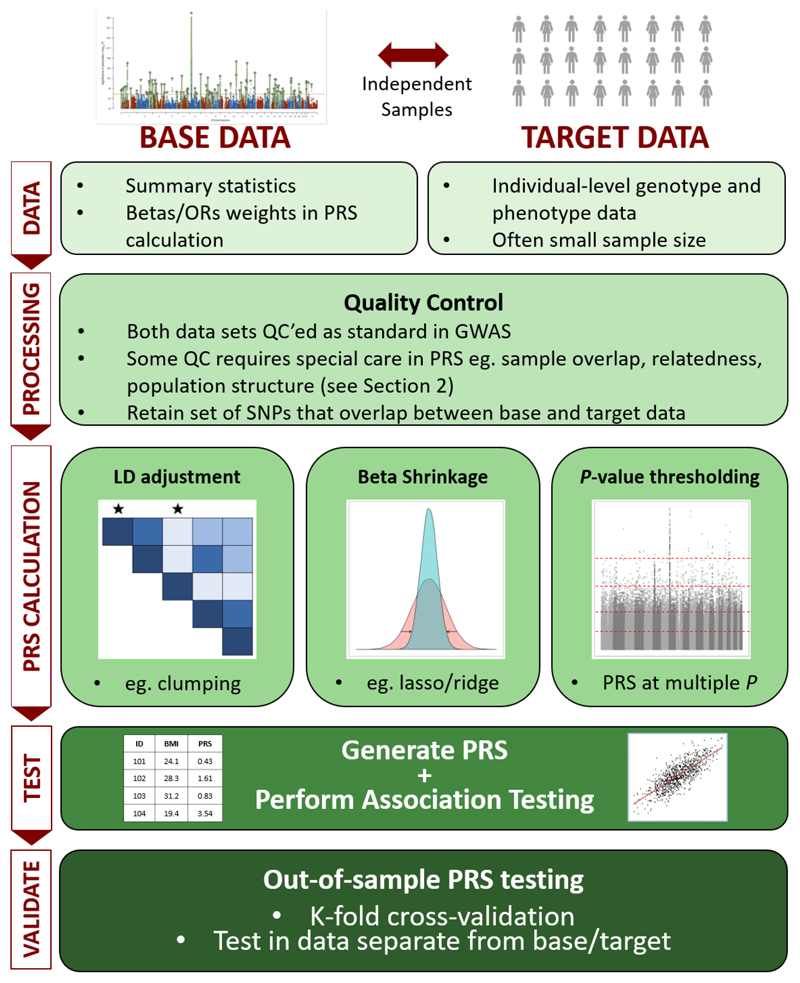
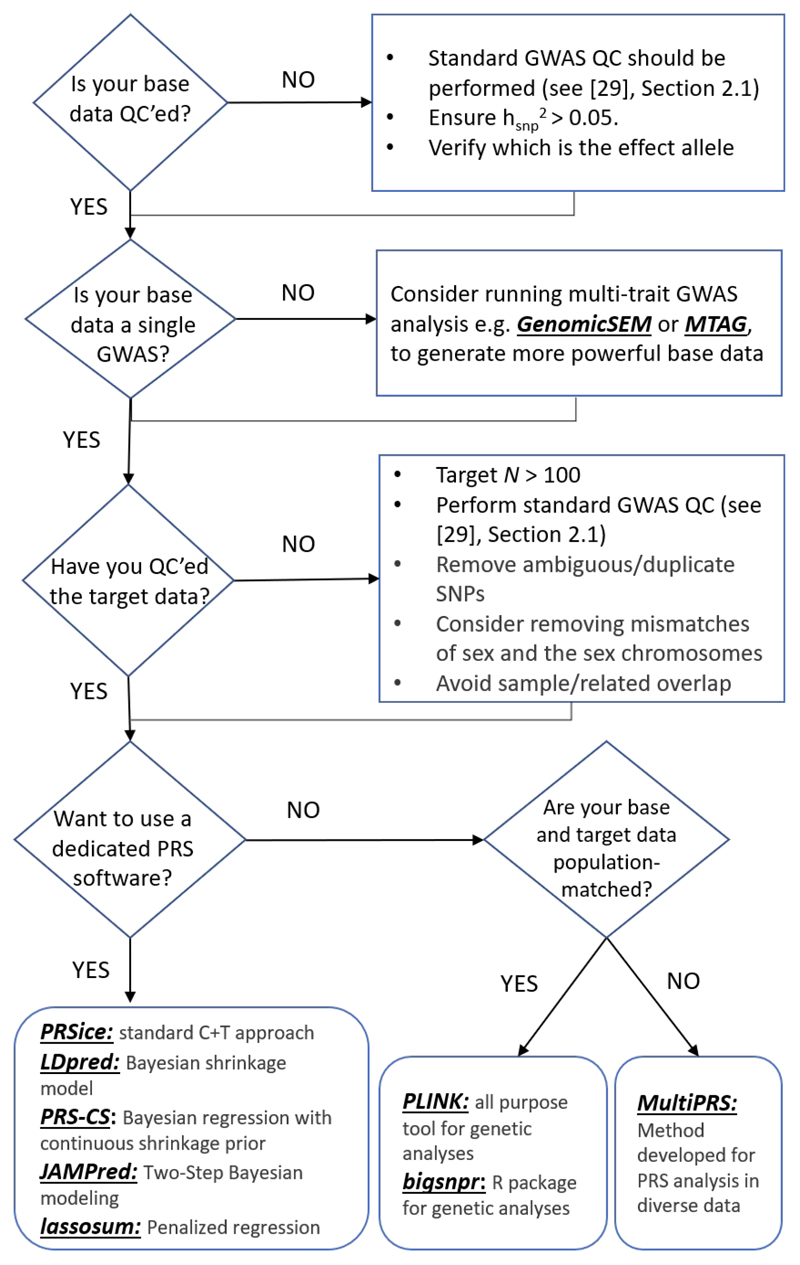
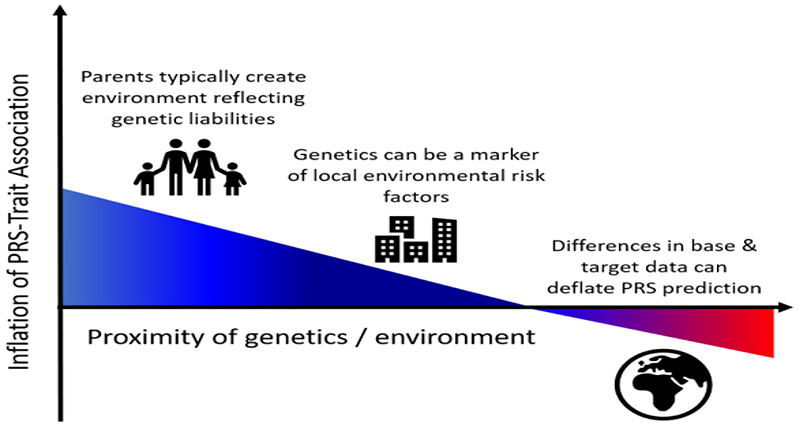
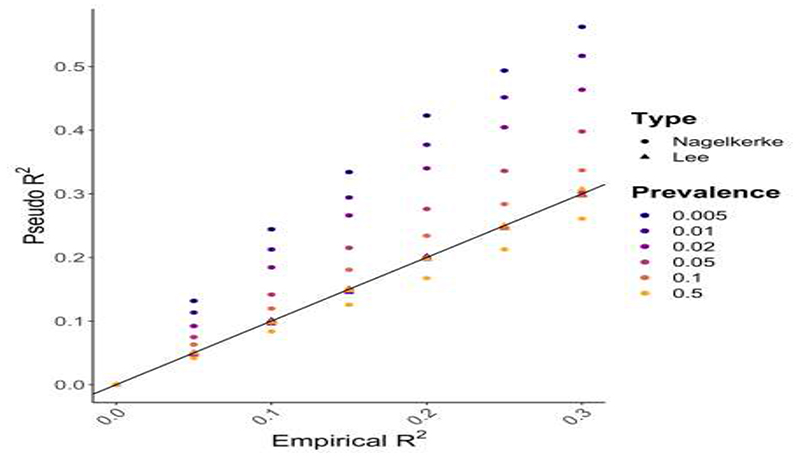
A polygenic score (PGS) or polygenic risk score (PRS) is an estimate of an individual's genetic liability to a trait or disease, calculated according to their genotype profile and relevant genome-wide association study (GWAS) data. While present PRSs typically explain only a small fraction of trait variance, their correlation with the single largest contributor to phenotypic variation-genetic liability-has led to the routine application of PRSs across biomedical research. Among a range of applications, PRSs are exploited to assess shared etiology between phenotypes, to evaluate the clinical utility of genetic data for complex disease and as part of experimental studies in which, for example, experiments are performed that compare outcomes (e.g., gene expression and cellular response to treatment) between individuals with low and high PRS values. As GWAS sample sizes increase and PRSs become more powerful, PRSs are set to play a key role in research and stratified medicine. However, despite the importance and growing application of PRSs, there are limited guidelines for performing PRS analyses, which can lead to inconsistency between studies and misinterpretation of results. Here, we provide detailed guidelines for performing and interpreting PRS analyses. We outline standard quality control steps, discuss different methods for the calculation of PRSs, provide an introductory online tutorial, highlight common misconceptions relating to PRS results, offer recommendations for best practice and discuss future challenges.
Figures
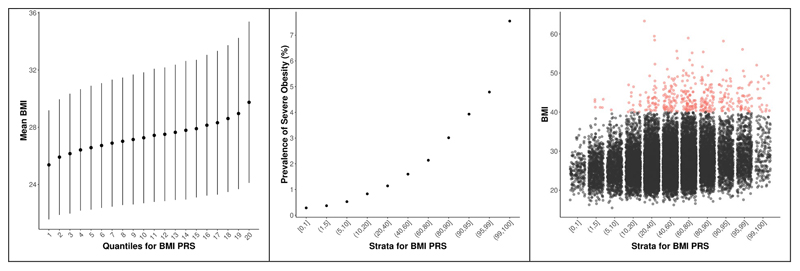
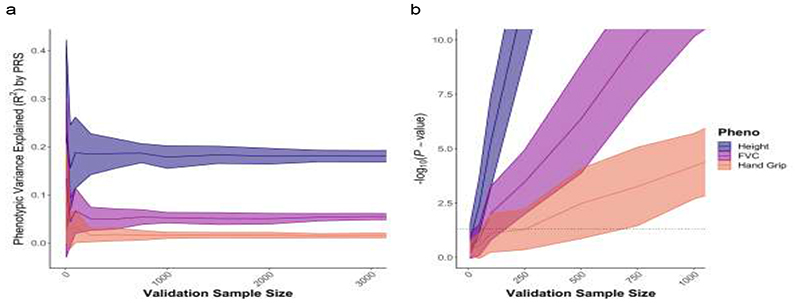
References
-
- Dudbridge F. Power and Predictive Accuracy of Polygenic Risk Scores. PLOS Genet. 2013;9:e1003348. [ A key theoretical PRS paper, providing the first analytical predictions of the performance of PRS analyses on real data. Formulae for computing expectations were derived according to factors such as trait heritability, base and target sample size, and polygenicity, assuming a quantitative genetics model. ] - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
