

Genetics of Atrial Fibrillation in 2020: GWAS, Genome Sequencing, Polygenic Risk, and Beyond
- PMID: 32716721
- PMCID: PMC7388073
- DOI: 10.1161/CIRCRESAHA.120.316575
Genetics of Atrial Fibrillation in 2020: GWAS, Genome Sequencing, Polygenic Risk, and Beyond
Abstract
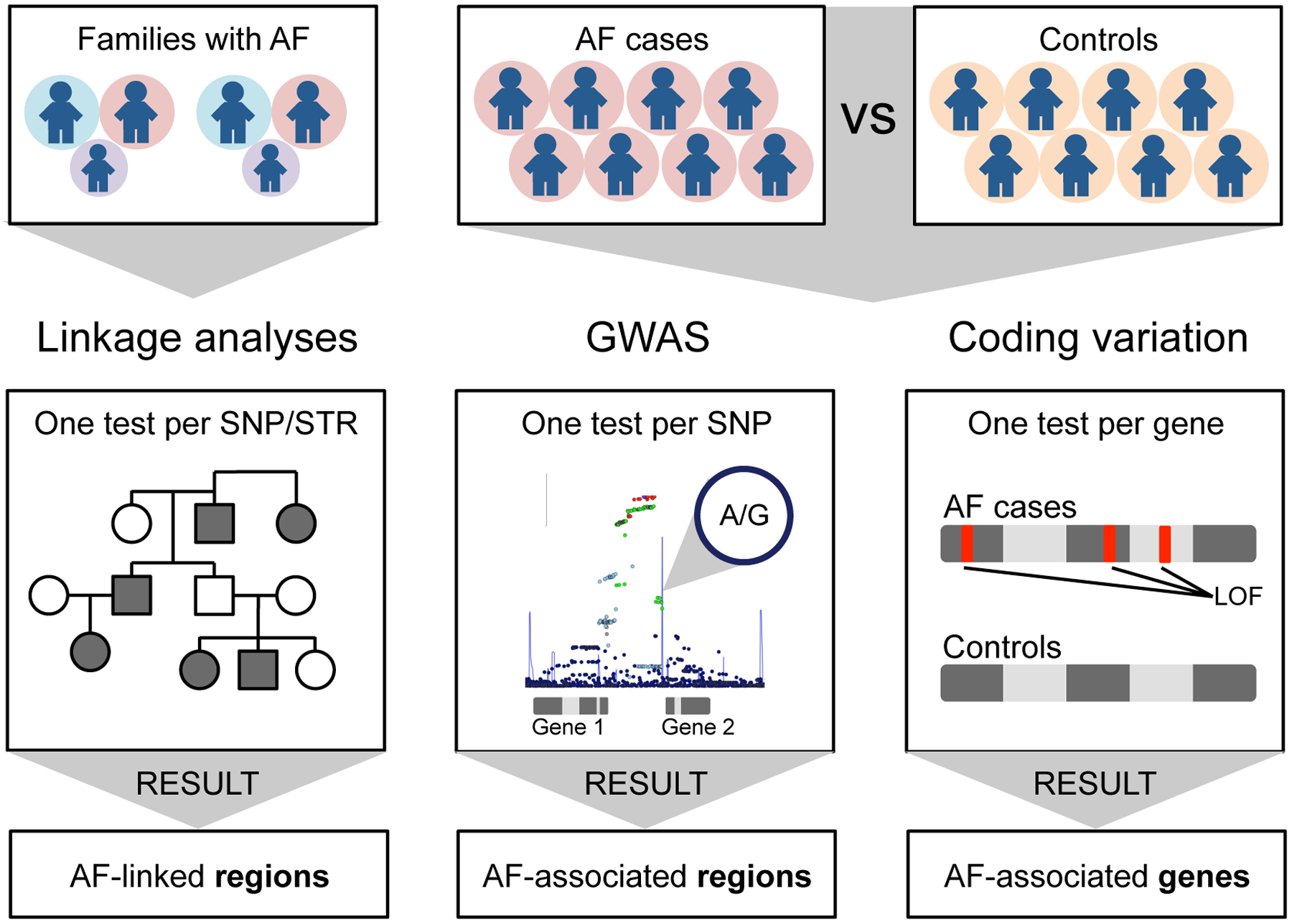
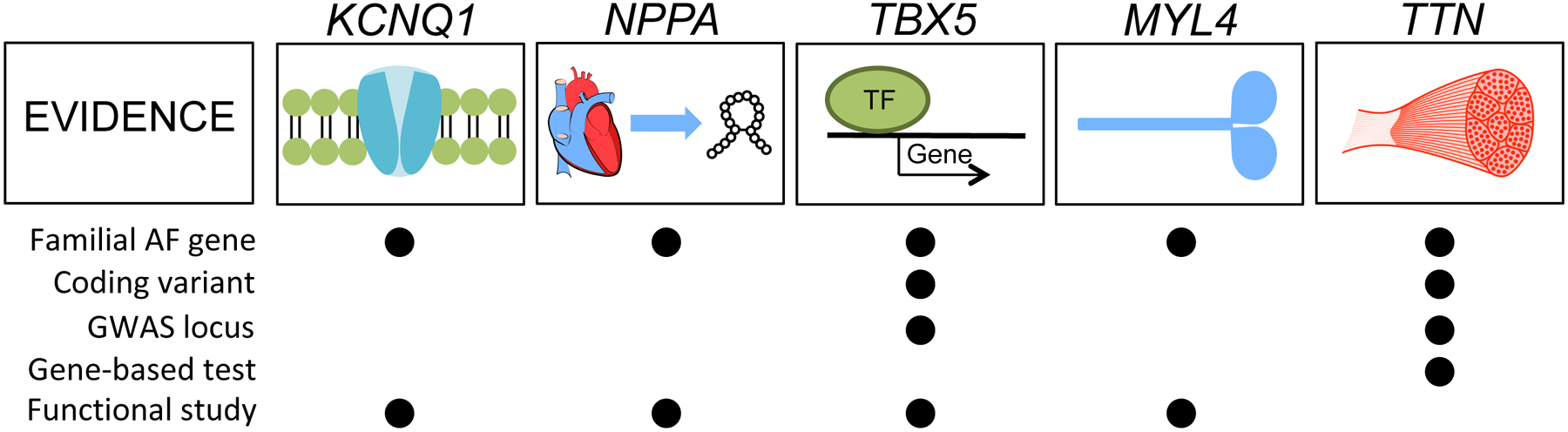
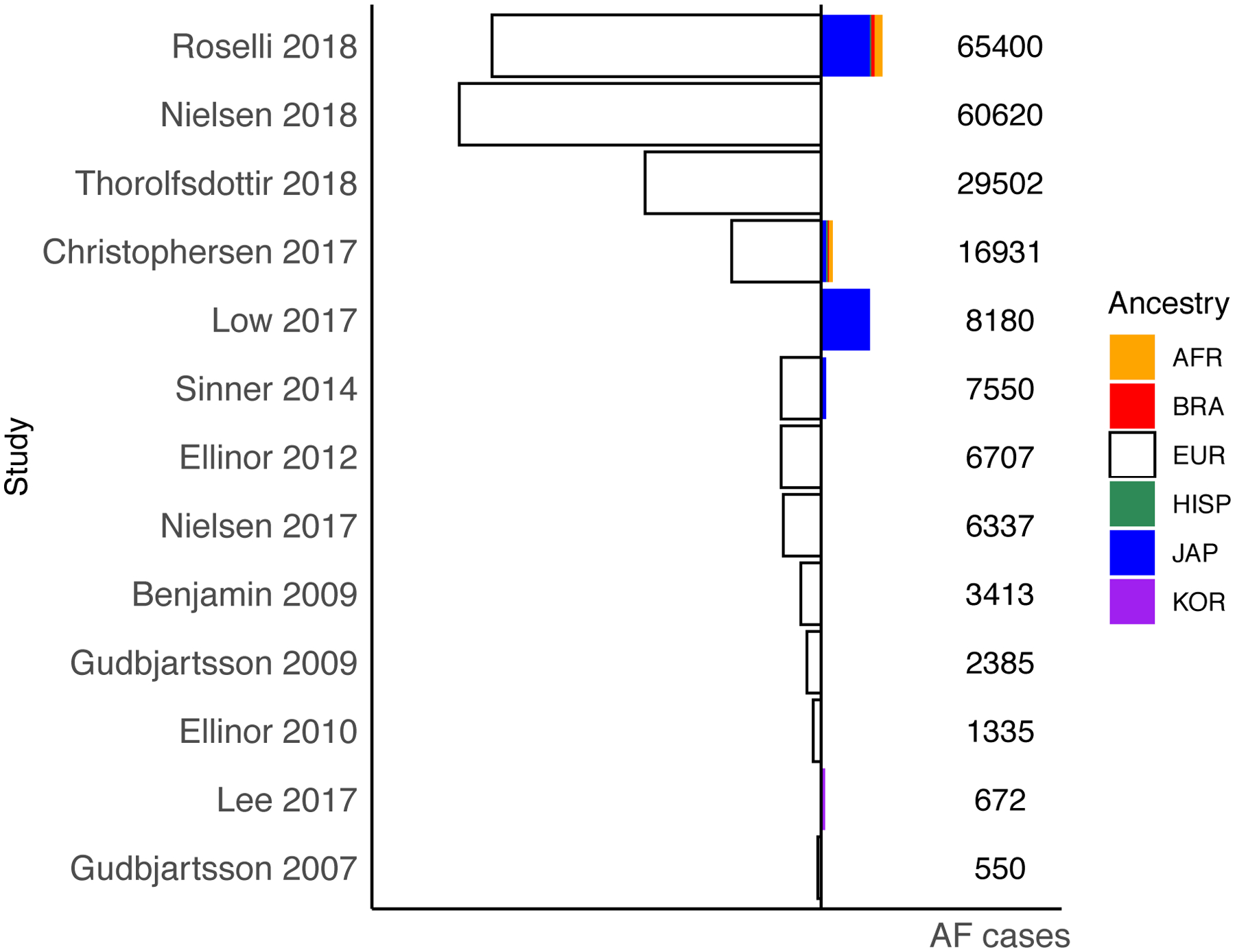
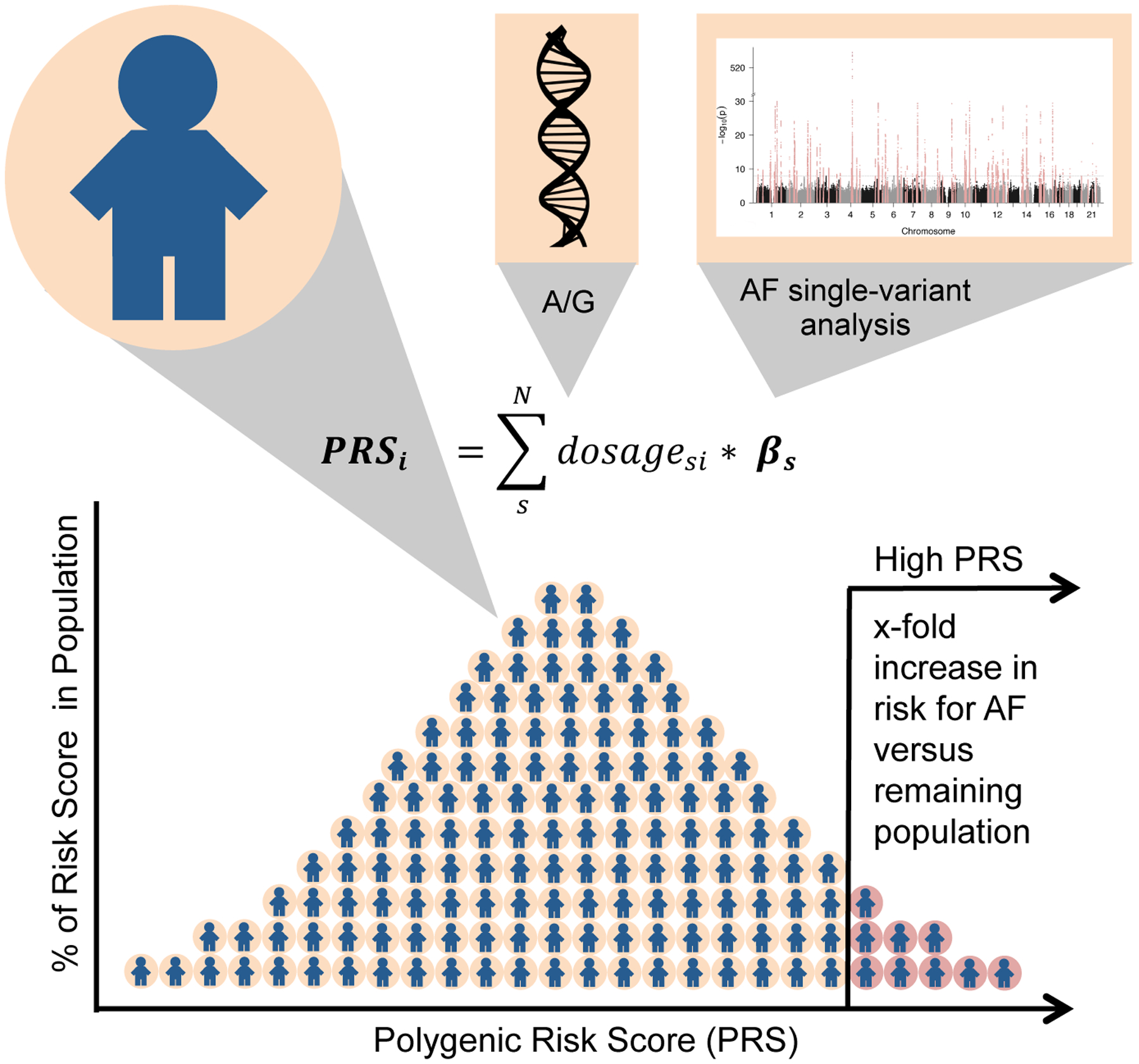
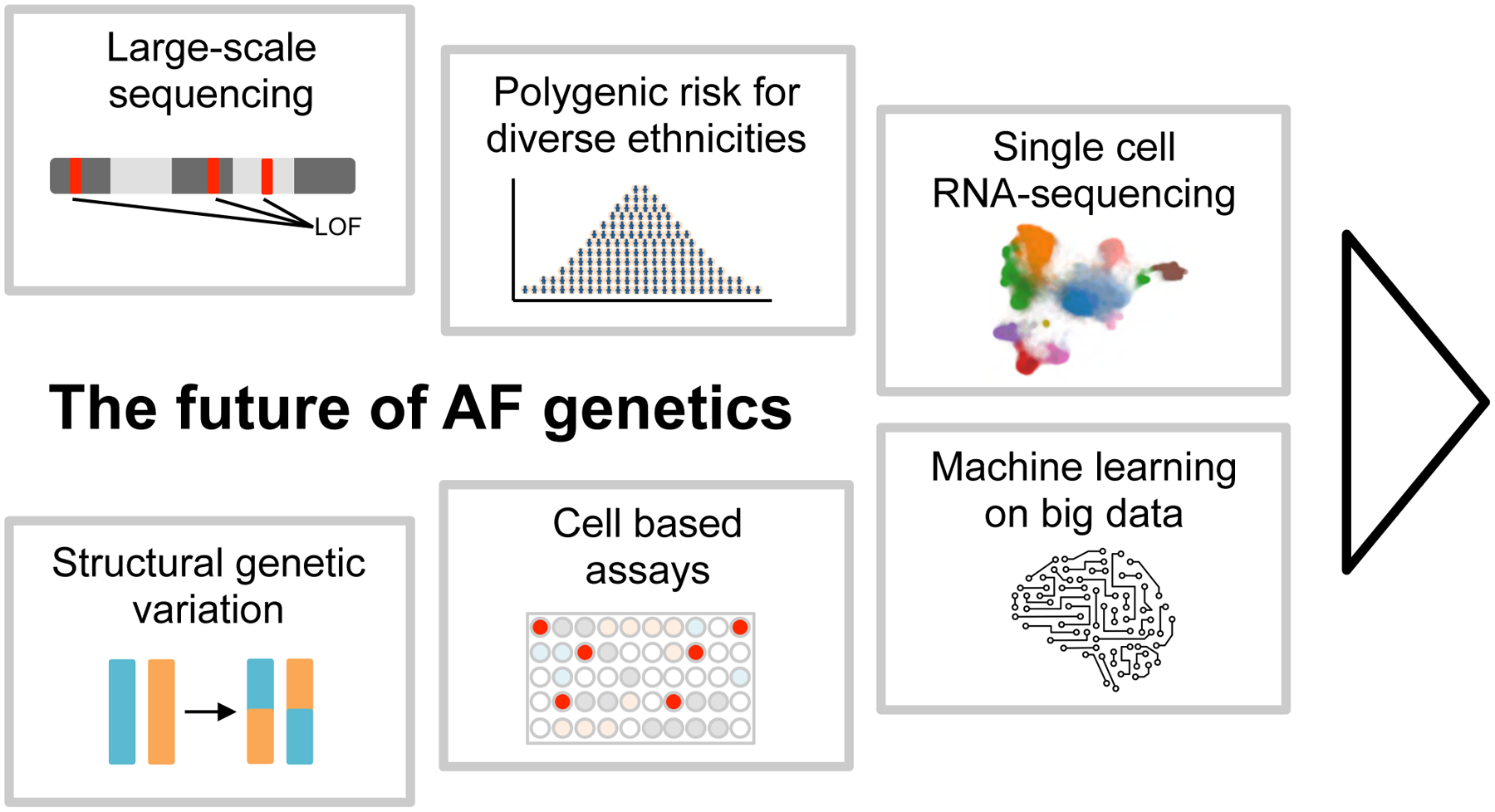
Atrial fibrillation is a common heart rhythm disorder that leads to an increased risk for stroke and heart failure. Atrial fibrillation is a complex disease with both environmental and genetic risk factors that contribute to the arrhythmia. Over the last decade, rapid progress has been made in identifying the genetic basis for this common condition. In this review, we provide an overview of the primary types of genetic analyses performed for atrial fibrillation, including linkage studies, genome-wide association studies, and studies of rare coding variation. With these results in mind, we aim to highlighting the existing knowledge gaps and future directions for atrial fibrillation genetics research.
Keywords: atrial fibrillation; exome; genetics; genome-wide association study; mutation.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
