

Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis
- PMID: 32717081
- PMCID: PMC7478737
- DOI: 10.1084/jem.20191699
Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis
Abstract
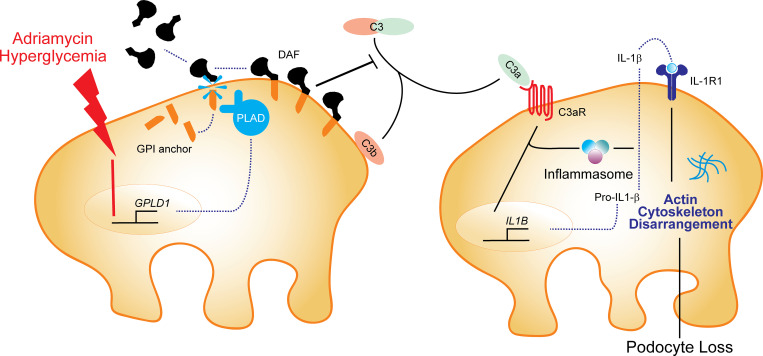
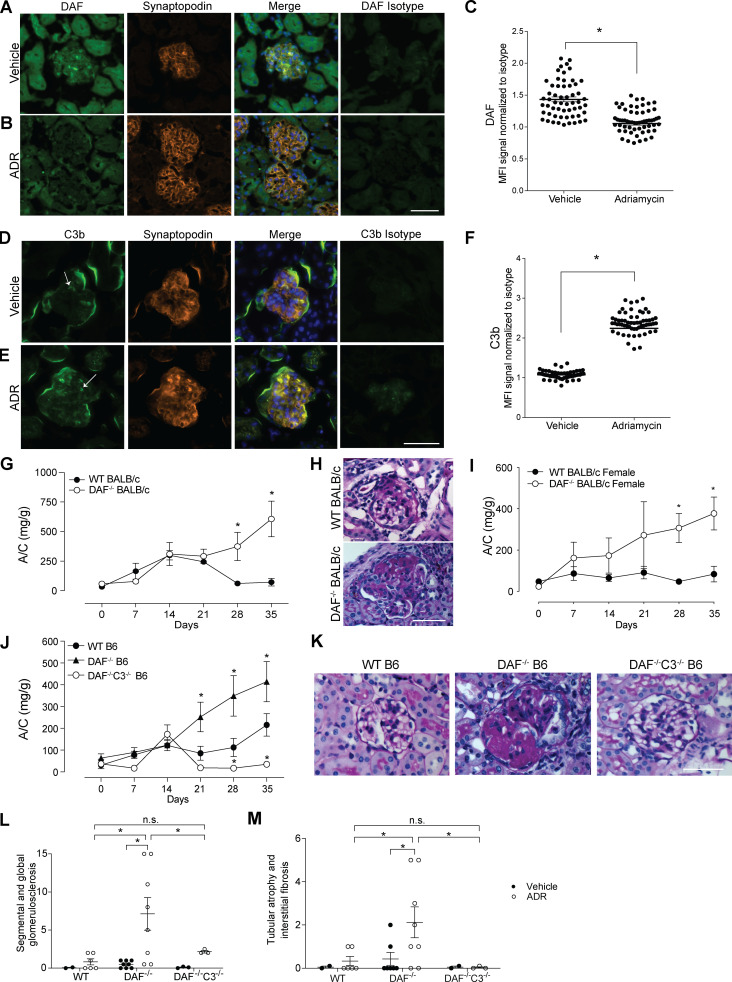
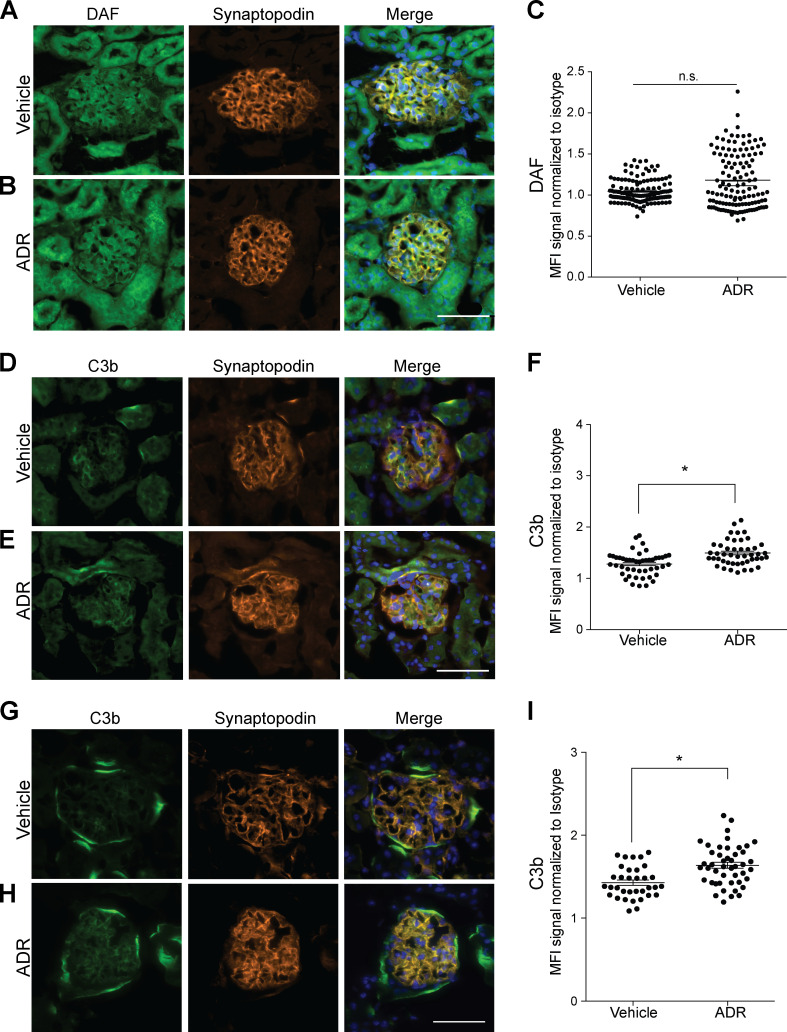
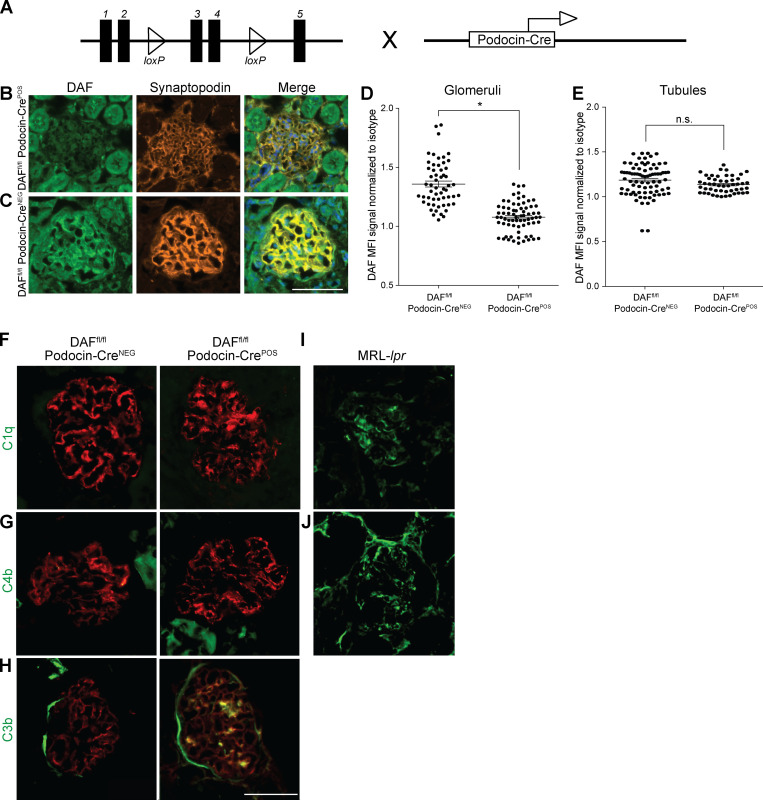
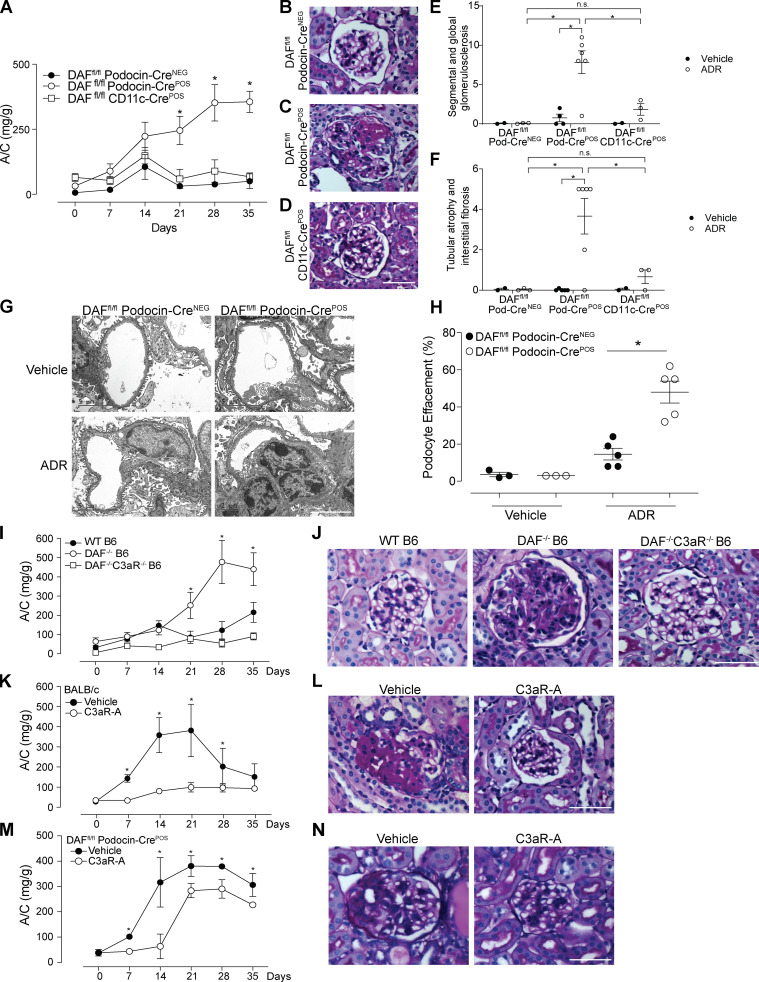
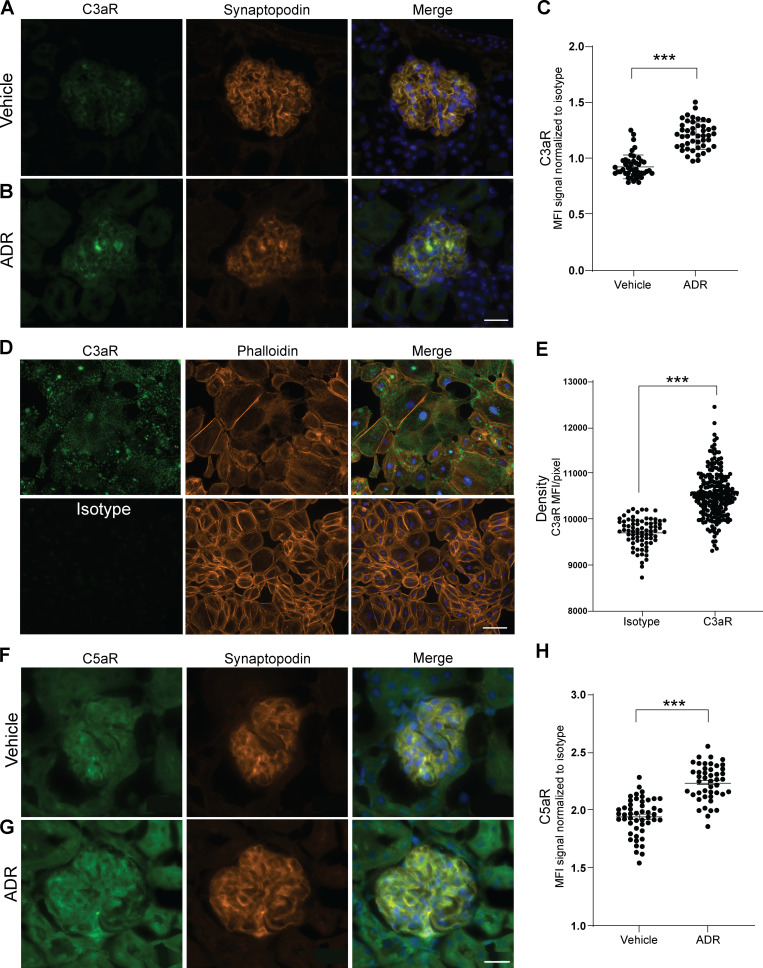
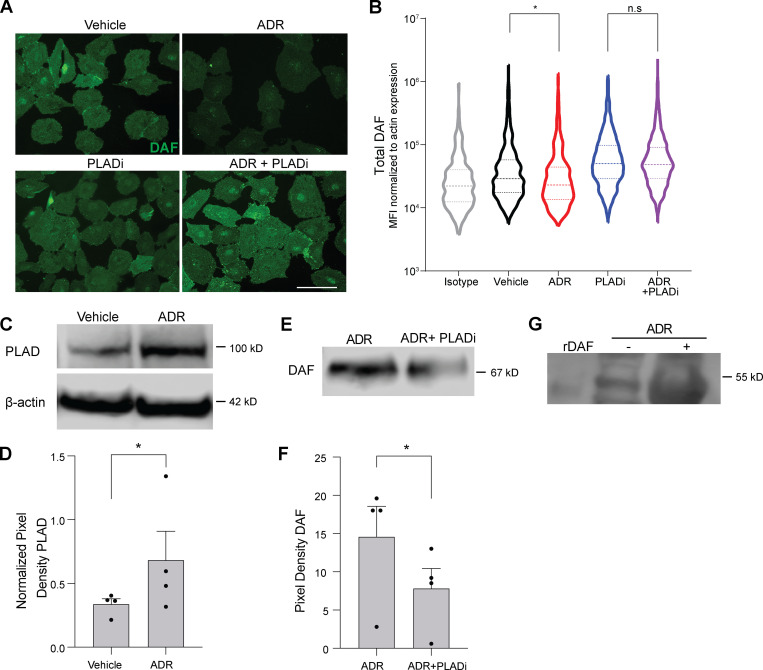
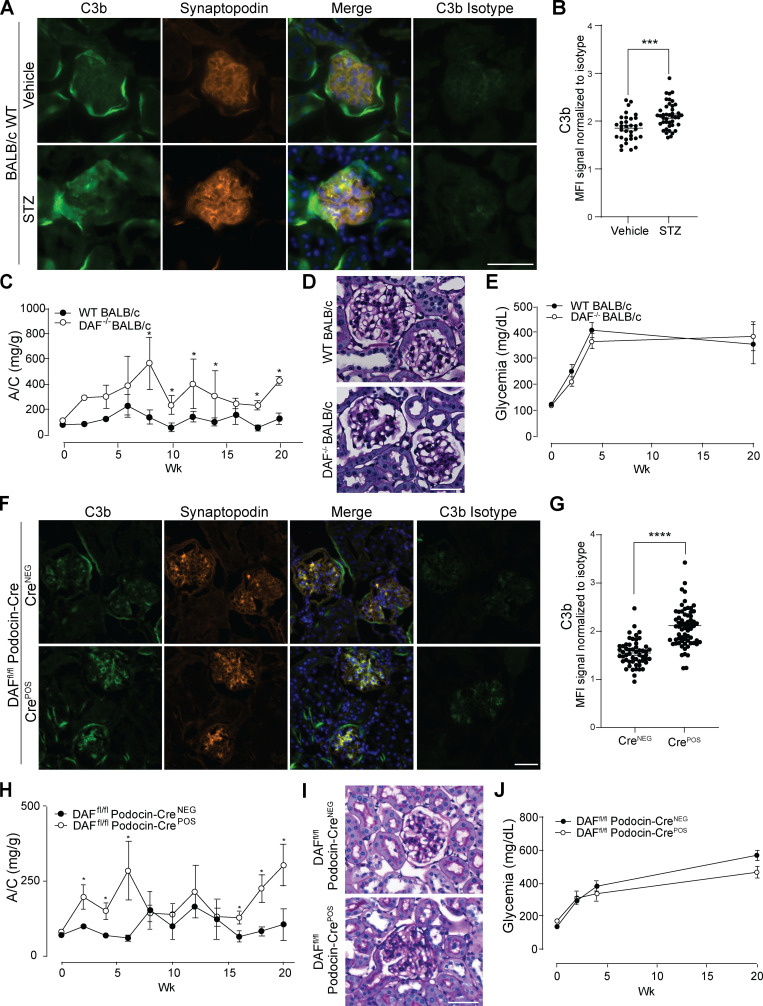
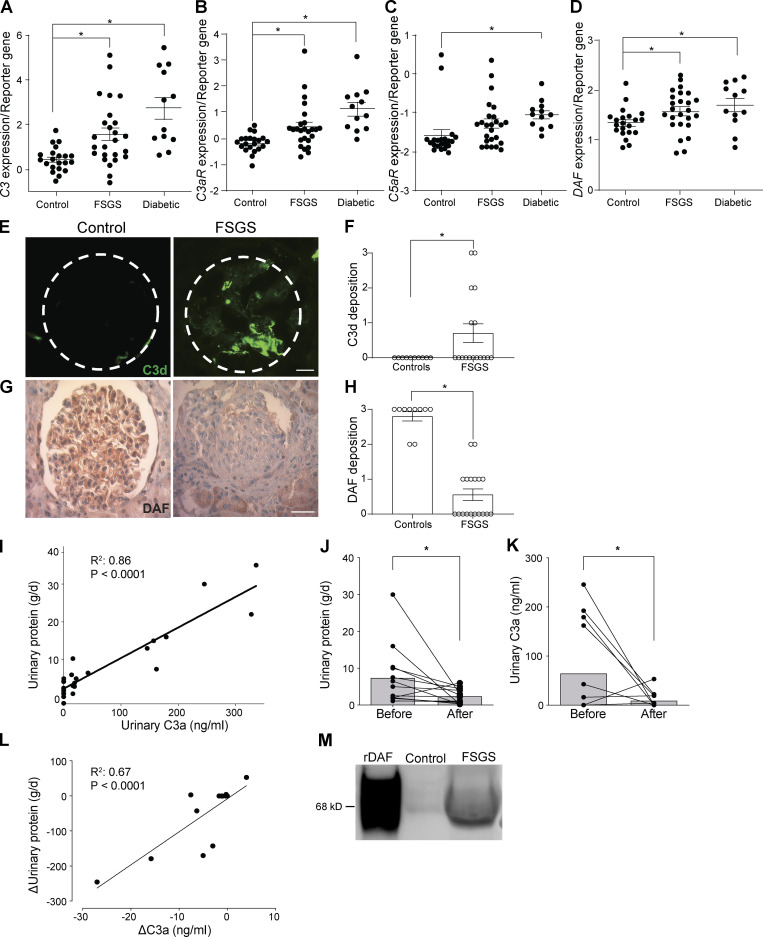
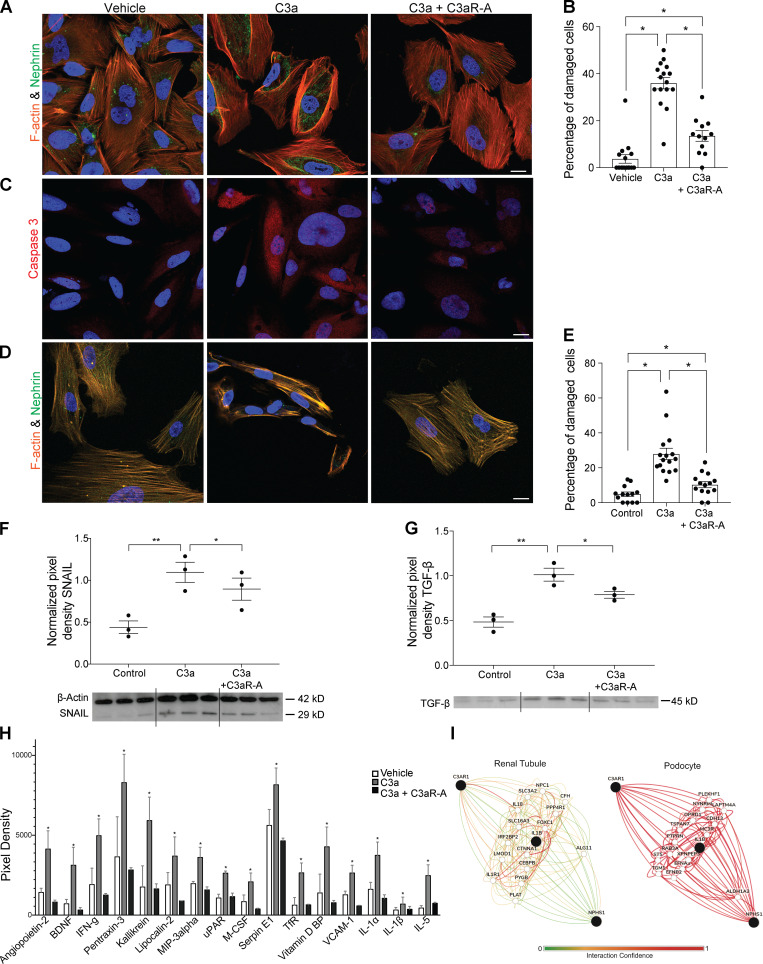
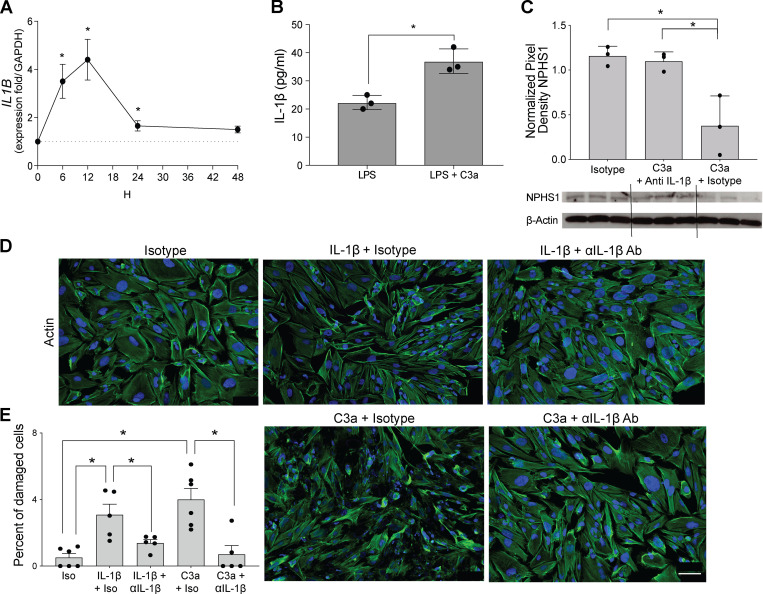
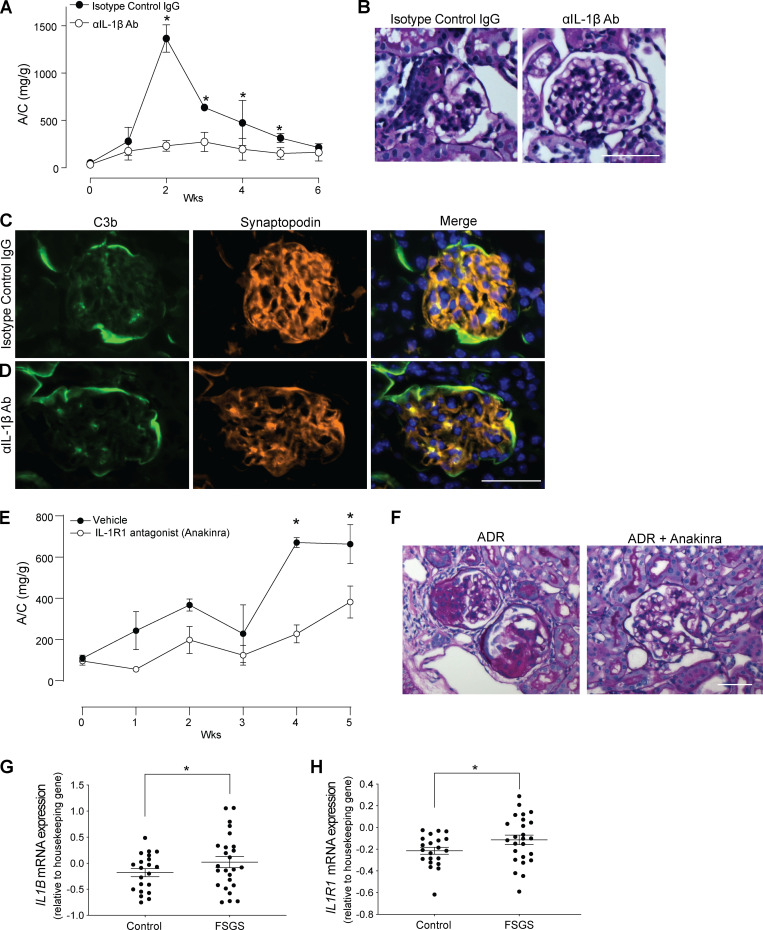
Kidney glomerulosclerosis commonly progresses to end-stage kidney failure, but pathogenic mechanisms are still poorly understood. Here, we show that podocyte expression of decay-accelerating factor (DAF/CD55), a complement C3 convertase regulator, crucially controls disease in murine models of adriamycin (ADR)-induced focal and segmental glomerulosclerosis (FSGS) and streptozotocin (STZ)-induced diabetic glomerulosclerosis. ADR induces enzymatic cleavage of DAF from podocyte surfaces, leading to complement activation. C3 deficiency or prevention of C3a receptor (C3aR) signaling abrogates disease despite DAF deficiency, confirming complement dependence. Mechanistic studies show that C3a/C3aR ligations on podocytes initiate an autocrine IL-1β/IL-1R1 signaling loop that reduces nephrin expression, causing actin cytoskeleton rearrangement. Uncoupling IL-1β/IL-1R1 signaling prevents disease, providing a causal link. Glomeruli of patients with FSGS lack DAF and stain positive for C3d, and urinary C3a positively correlates with the degree of proteinuria. Together, our data indicate that the development and progression of glomerulosclerosis involve loss of podocyte DAF, triggering local, complement-dependent, IL-1β-induced podocyte injury, potentially identifying new therapeutic targets.
© 2020 Angeletti et al.
Conflict of interest statement
Disclosures: J.M. Thurman reported a patent to US 7,999,082 issued "Alexion Pharmaceuticals, Inc." and a patent to US 8,703,140 B2 issued "Alexion Pharmaceuticals, Inc." J. Manrique reported personal fees from Alexion Pharmaceuticals, Inc. P.S. Heeger reported personal fees from Mallinckrodt Pharmaceuticals during the conduct of the study. No other disclosures were reported.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
