

Identification of high-affinity inhibitors of SARS-CoV-2 main protease: Towards the development of effective COVID-19 therapy
- PMID: 32717346
- PMCID: PMC7380256
- DOI: 10.1016/j.virusres.2020.198102
Identification of high-affinity inhibitors of SARS-CoV-2 main protease: Towards the development of effective COVID-19 therapy
Abstract
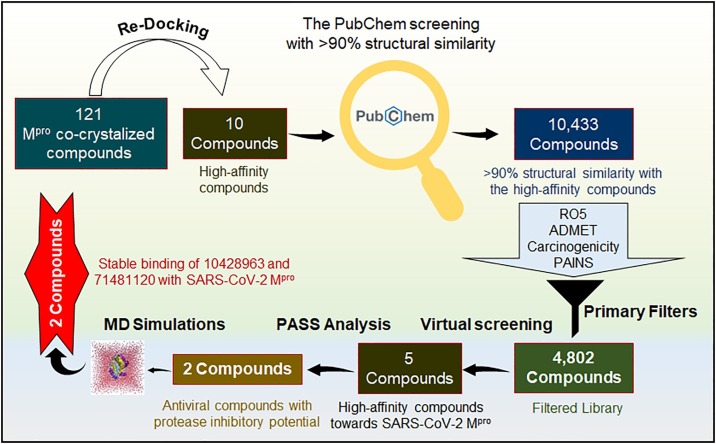
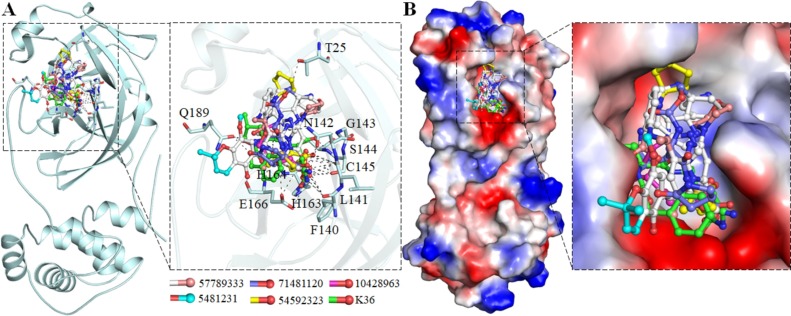
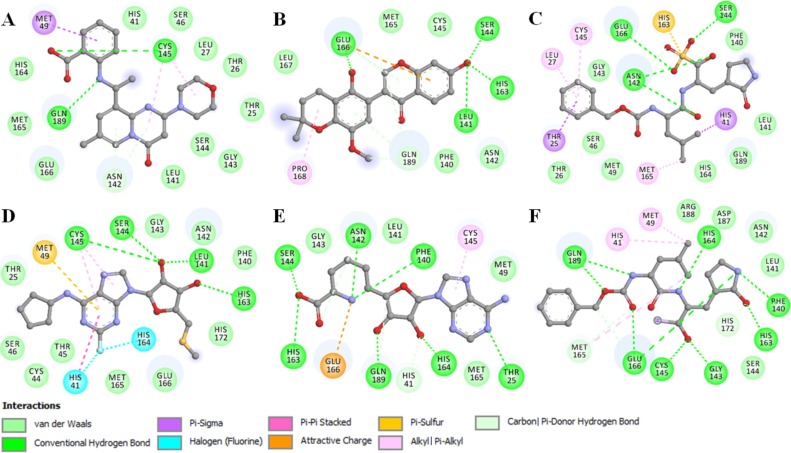
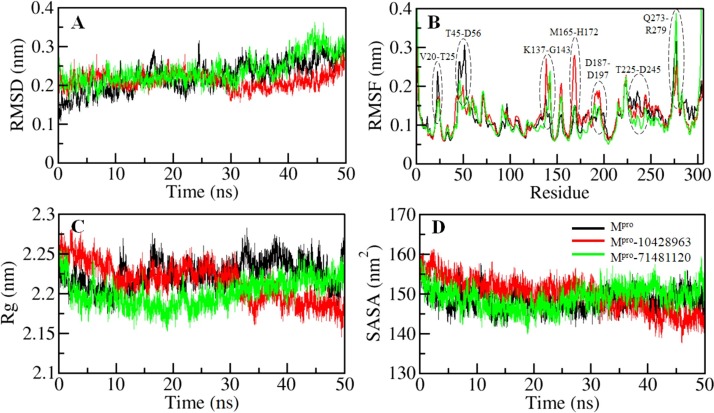
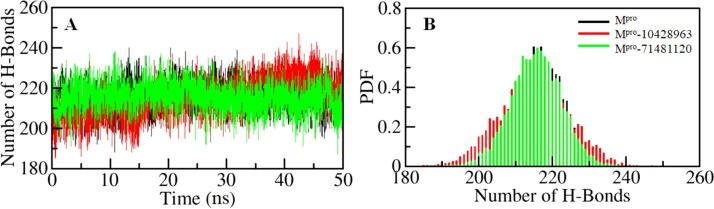
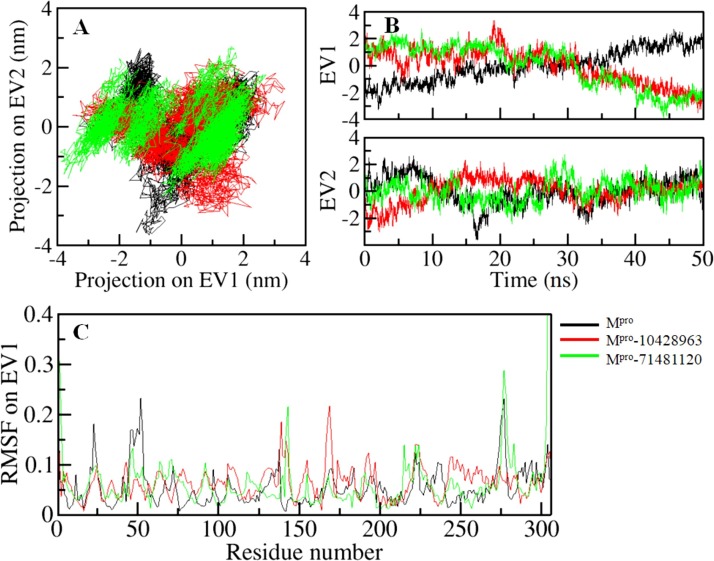
Coronavirus disease 2019 (COVID-19) is an infectious disease, caused by a newly emerged highly pathogenic virus called novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Targeting the main protease (Mpro, 3CLpro) of SARS-CoV-2 is an appealing approach for drug development because this enzyme plays a significant role in the viral replication and transcription. The available crystal structures of SARS-CoV-2 Mpro determined in the presence of different ligands and inhibitor-like compounds provide a platform for the quick development of selective inhibitors of SARS-CoV-2 Mpro. In this study, we utilized the structural information of co-crystallized SARS-CoV-2 Mpro for the structure-guided drug discovery of high-affinity inhibitors from the PubChem database. The screened compounds were selected on the basis of their physicochemical properties, drug-likeliness, and strength of affinity to the SARS-CoV-2 Mpro. Finally, we have identified 6-Deaminosinefungin (PubChem ID: 10428963) and UNII-O9H5KY11SV (PubChem ID: 71481120) as potential inhibitors of SARS-CoV-2 Mpro which may be further exploited in drug development to address SARS-CoV-2 pathogenesis. Both compounds are structural analogs of known antivirals, having considerable protease inhibitory potential with improved pharmacological properties. All-atom molecular dynamics simulations suggested SARS-CoV-2 Mpro in complex with these compounds is stable during the simulation period with minimal structural changes. This work provides enough evidence for further implementation of the identified compounds in the development of effective therapeutics of COVID-19.
Keywords: Coronavirus disease 2019; Drug discovery; Main protease; Molecular dynamics simulations; SARS-CoV-2; Virtual screening.
Copyright © 2020 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
-
- Altis A., Otten M., Nguyen P.H., Hegger R., Stock G. Construction of the free energy landscape of biomolecules via dihedral angle principal component analysis. J. Chem. Phys. 2008;128 - PubMed
-
- Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993;17:412–425. - PubMed
-
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. - PubMed
-
- Ausaf Ali S., Hassan I., Islam A., Ahmad F. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014;15:456–476. - PubMed
-
- Biovia D.S. Dassault Systèmes; San Diego: 2015. Discovery Studio Modeling Environment.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
