

Genome-wide detection of tandem DNA repeats that are expanded in autism
- PMID: 32717741
- PMCID: PMC9348607
- DOI: 10.1038/s41586-020-2579-z
Genome-wide detection of tandem DNA repeats that are expanded in autism
Abstract
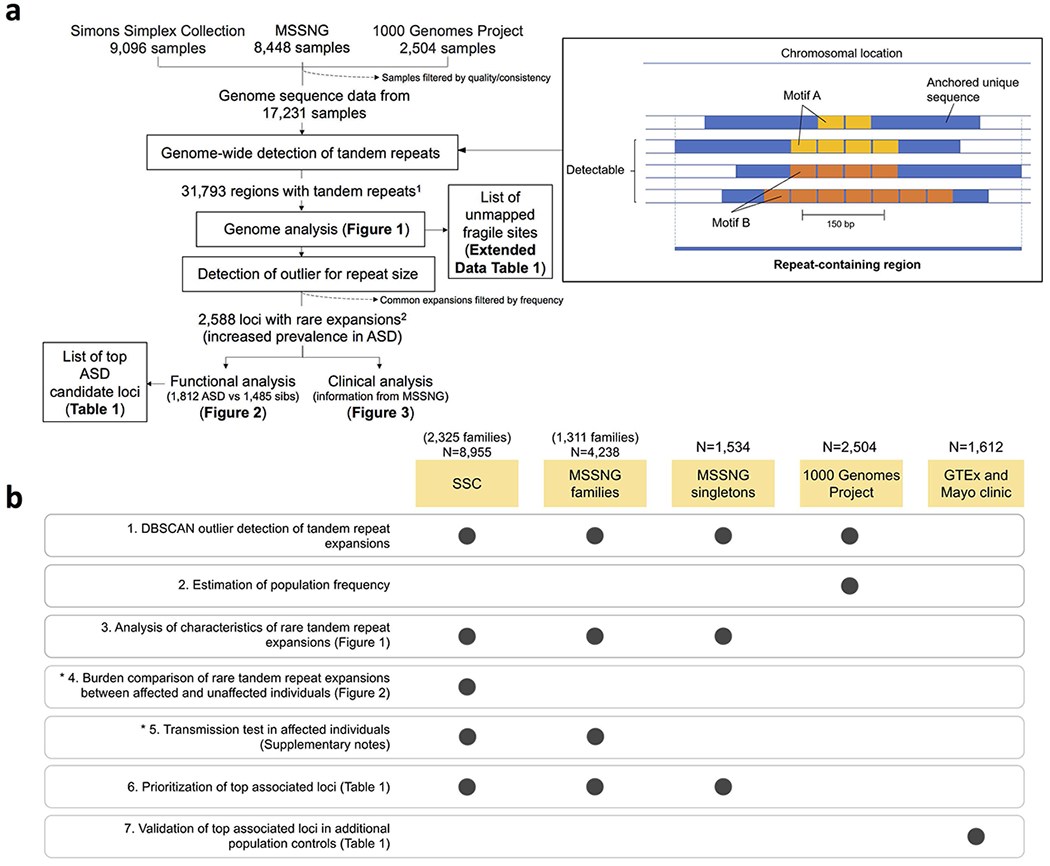
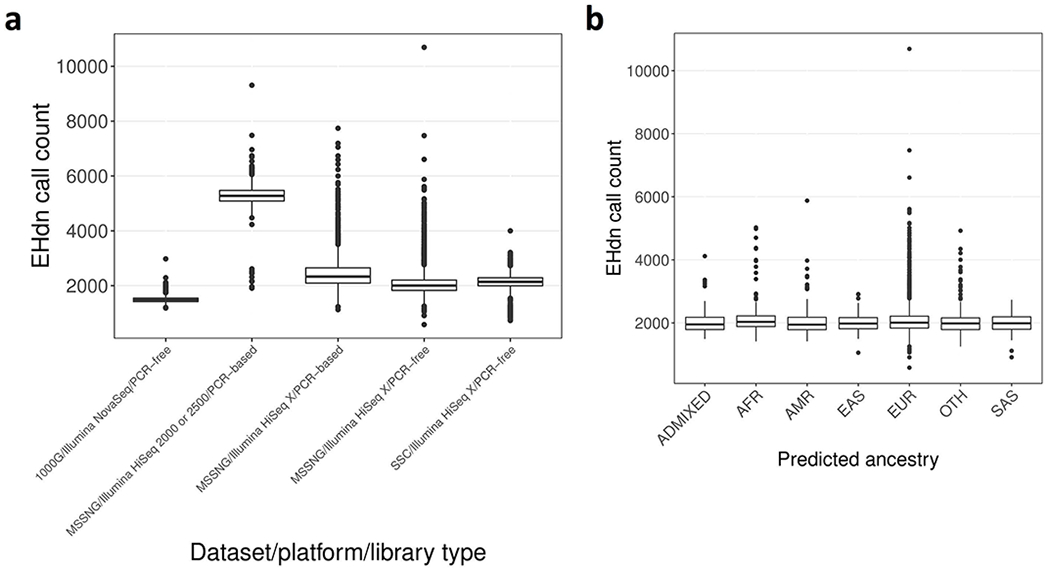
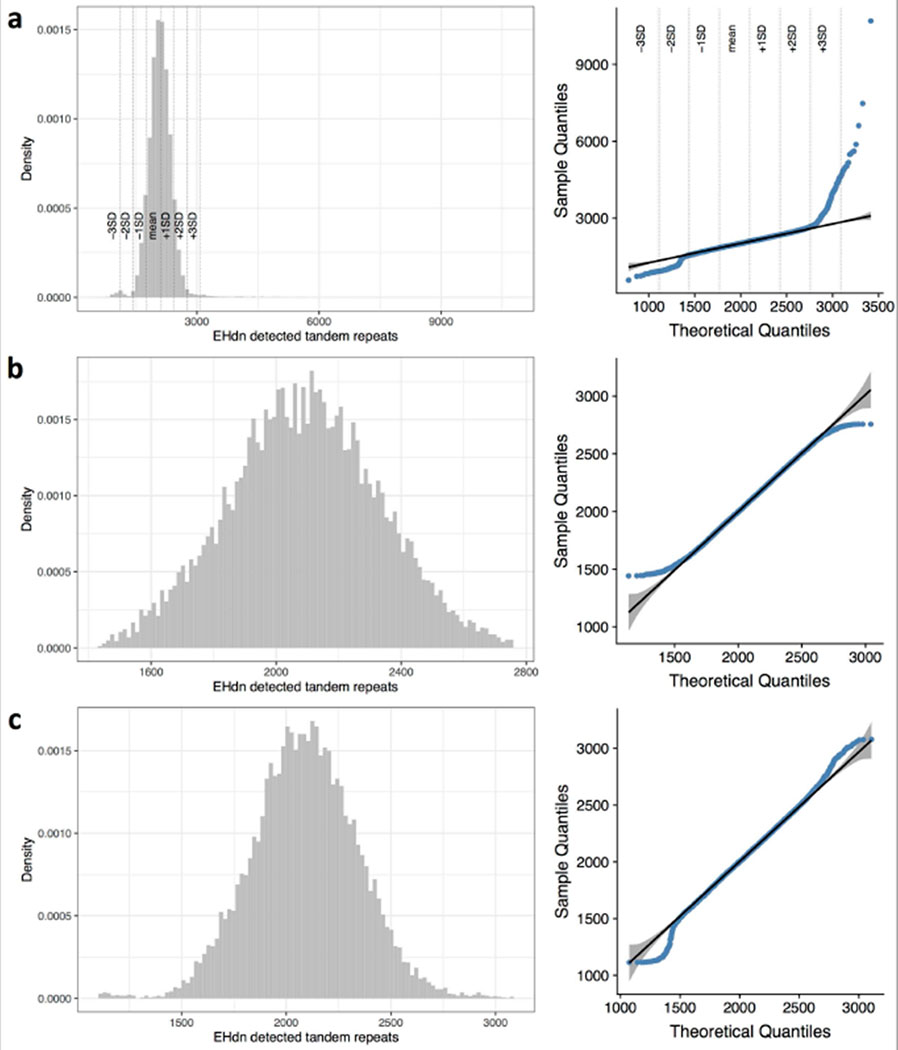
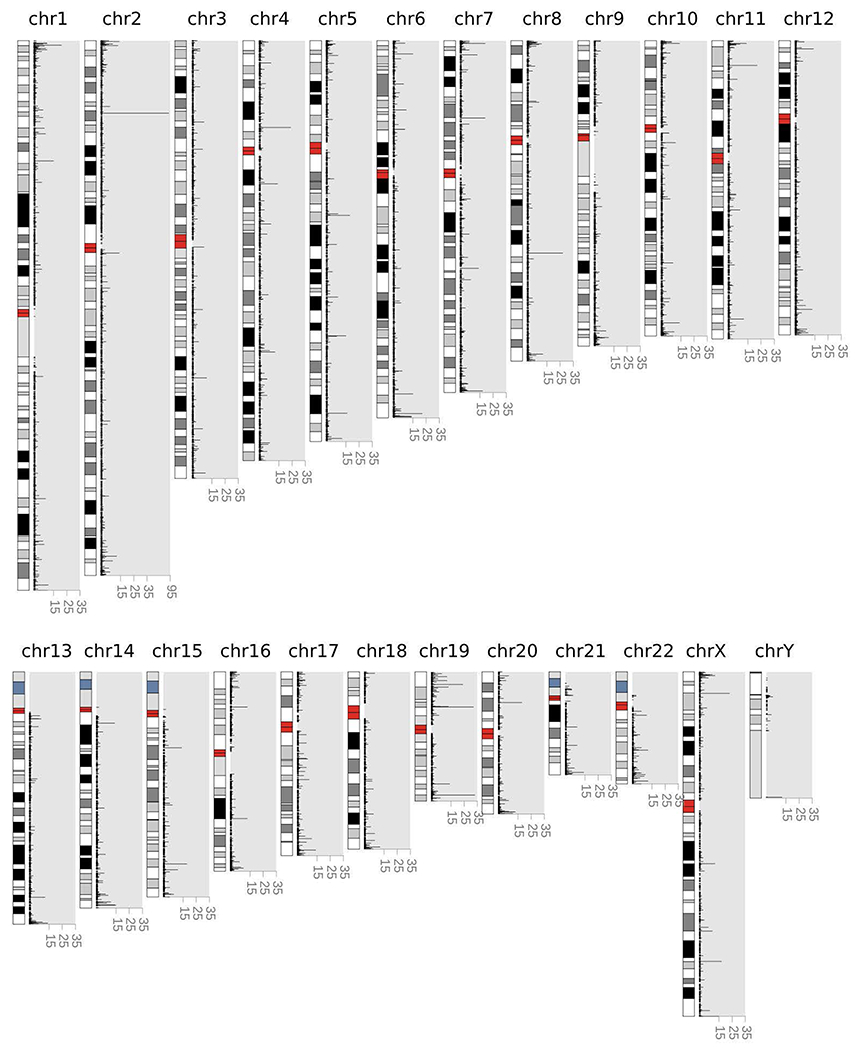
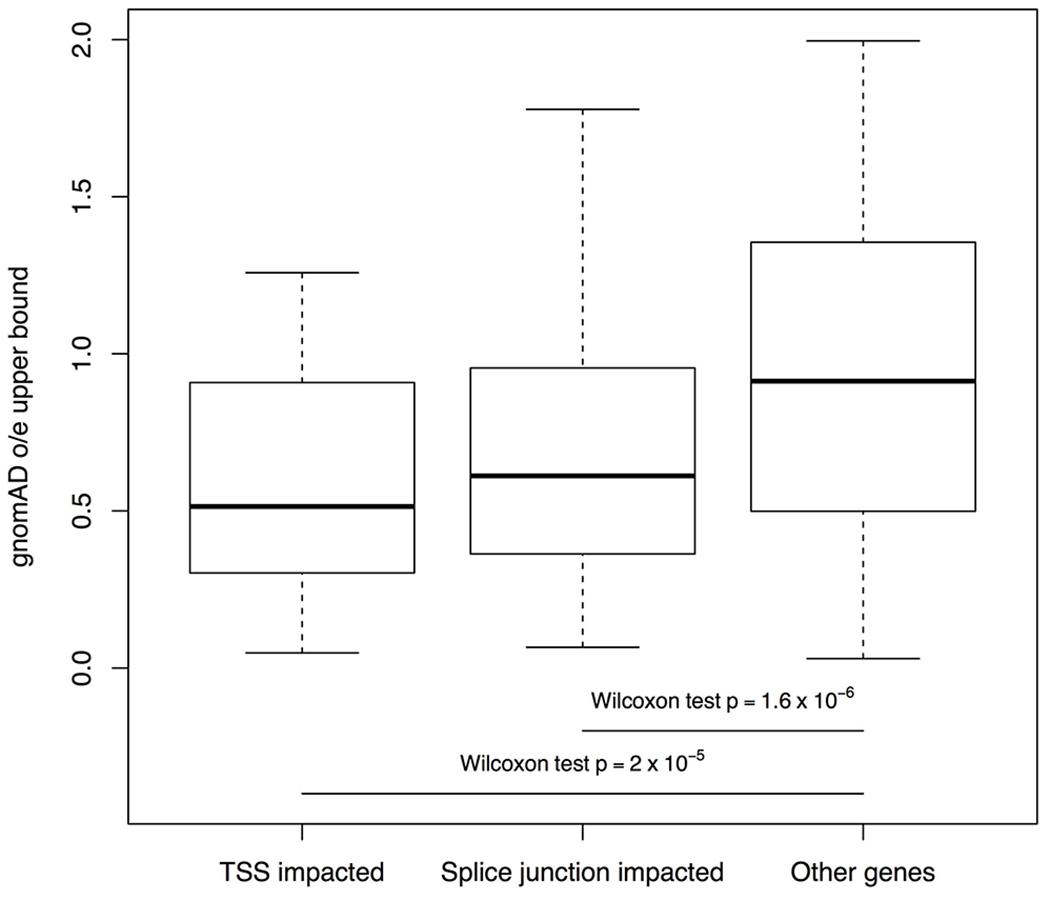
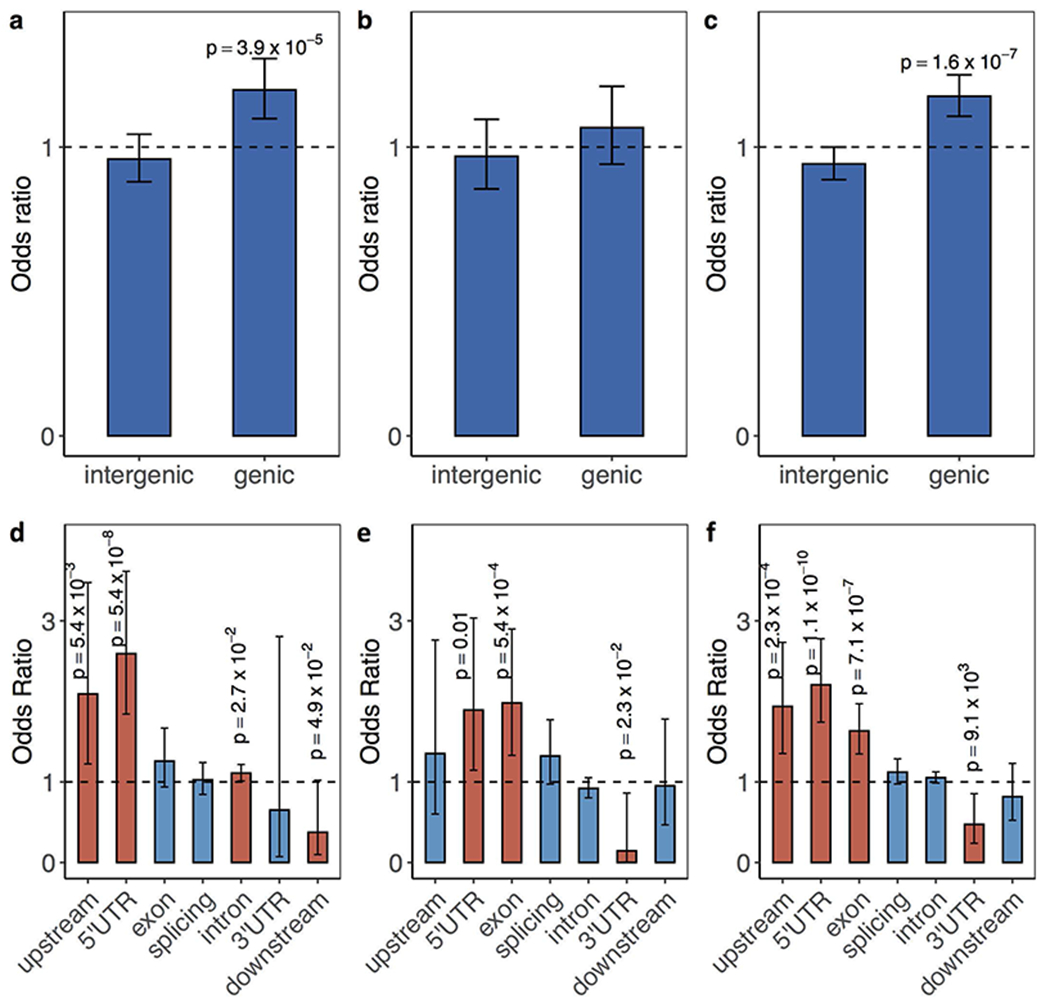
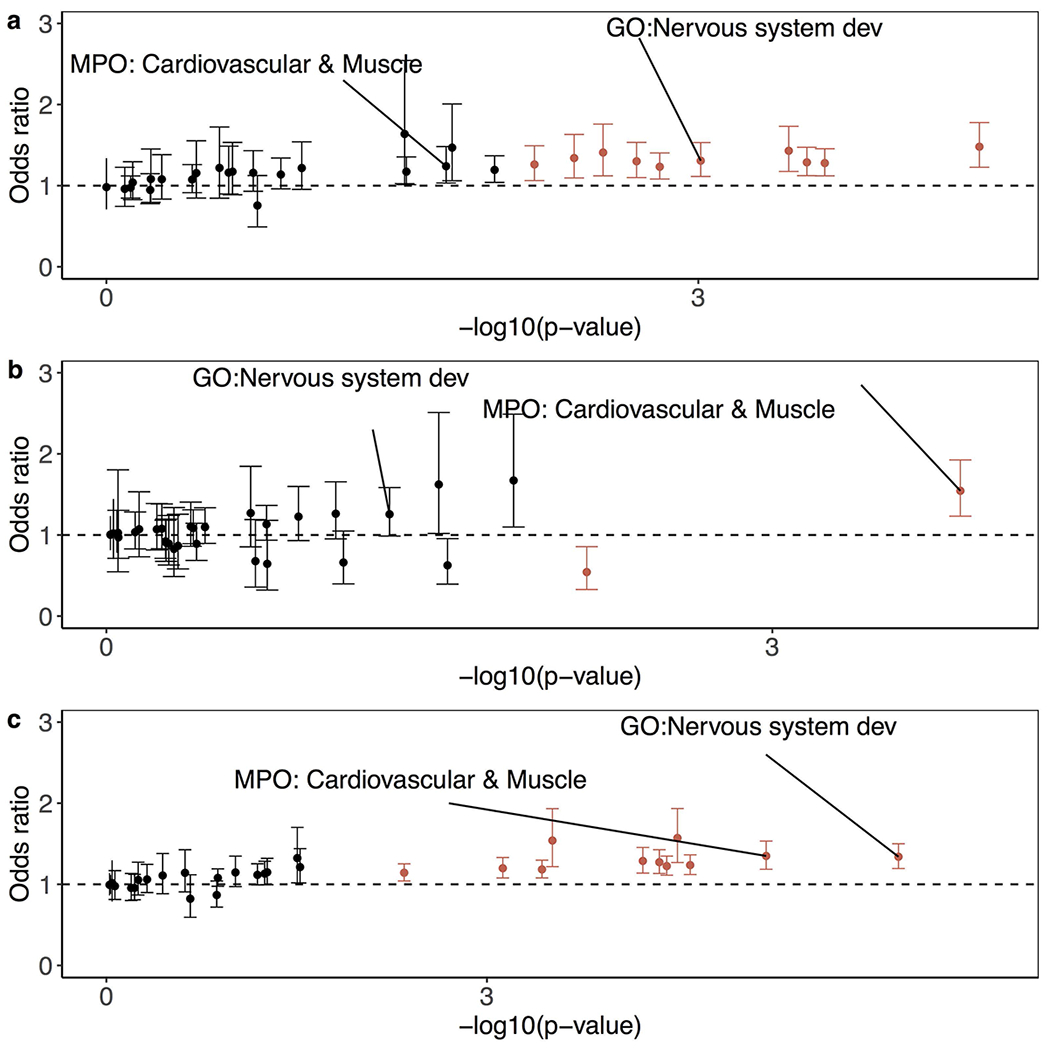
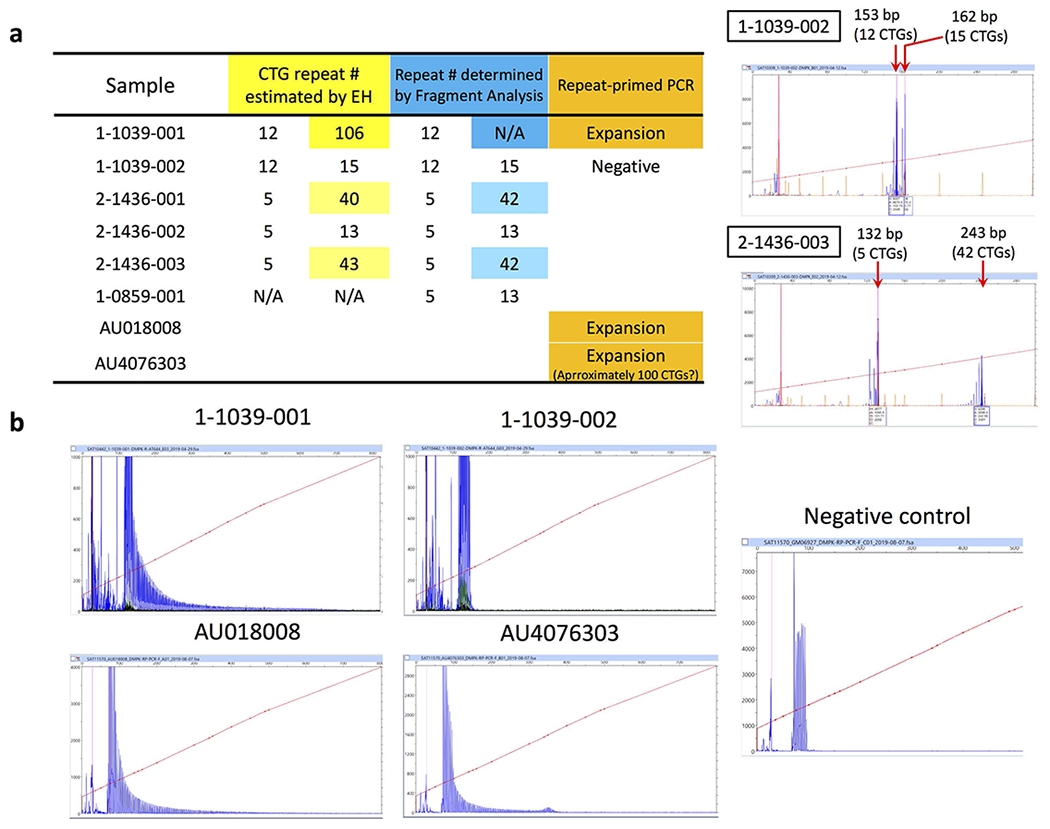
Tandem DNA repeats vary in the size and sequence of each unit (motif). When expanded, these tandem DNA repeats have been associated with more than 40 monogenic disorders1. Their involvement in disorders with complex genetics is largely unknown, as is the extent of their heterogeneity. Here we investigated the genome-wide characteristics of tandem repeats that had motifs with a length of 2-20 base pairs in 17,231 genomes of families containing individuals with autism spectrum disorder (ASD)2,3 and population control individuals4. We found extensive polymorphism in the size and sequence of motifs. Many of the tandem repeat loci that we detected correlated with cytogenetic fragile sites. At 2,588 loci, gene-associated expansions of tandem repeats that were rare among population control individuals were significantly more prevalent among individuals with ASD than their siblings without ASD, particularly in exons and near splice junctions, and in genes related to the development of the nervous system and cardiovascular system or muscle. Rare tandem repeat expansions had a prevalence of 23.3% in children with ASD compared with 20.7% in children without ASD, which suggests that tandem repeat expansions make a collective contribution to the risk of ASD of 2.6%. These rare tandem repeat expansions included previously undescribed ASD-linked expansions in DMPK and FXN, which are associated with neuromuscular conditions, and in previously unknown loci such as FGF14 and CACNB1. Rare tandem repeat expansions were associated with lower IQ and adaptive ability. Our results show that tandem DNA repeat expansions contribute strongly to the genetic aetiology and phenotypic complexity of ASD.
Figures
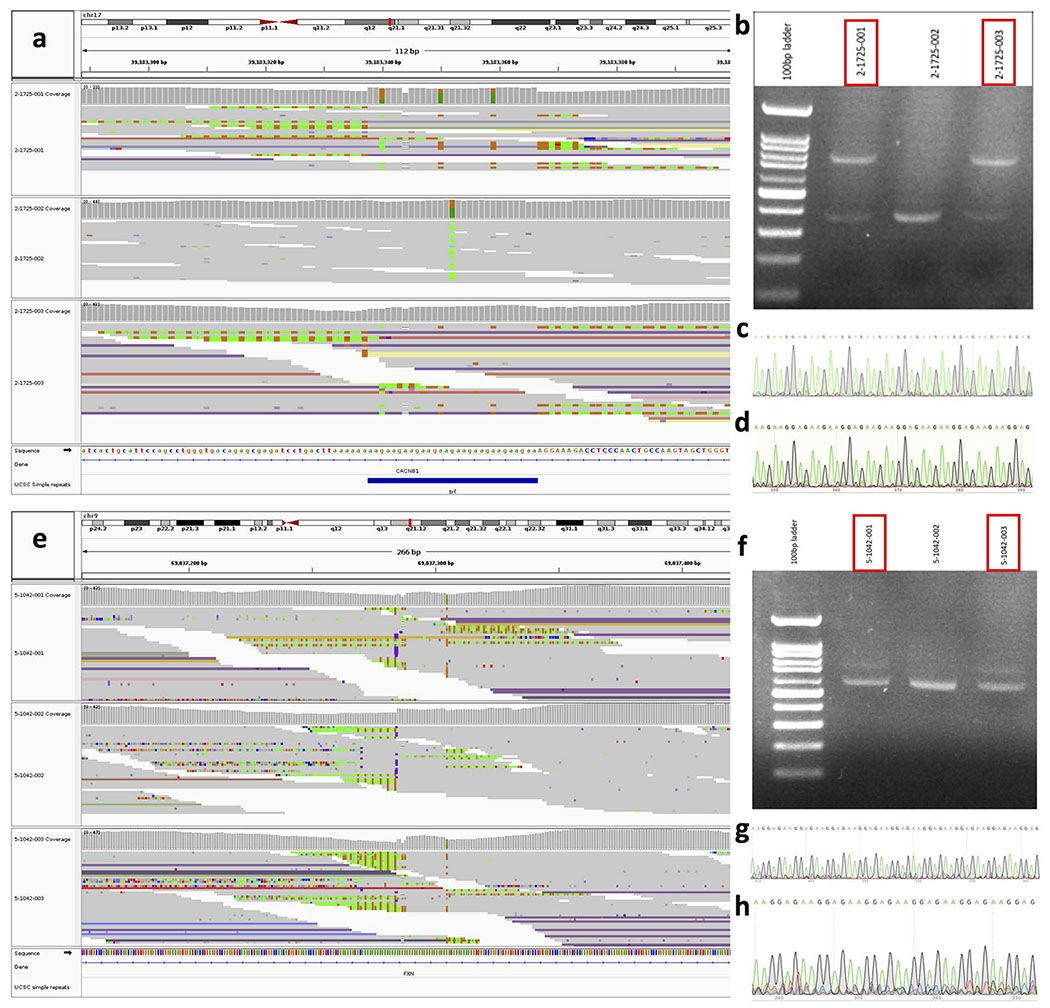
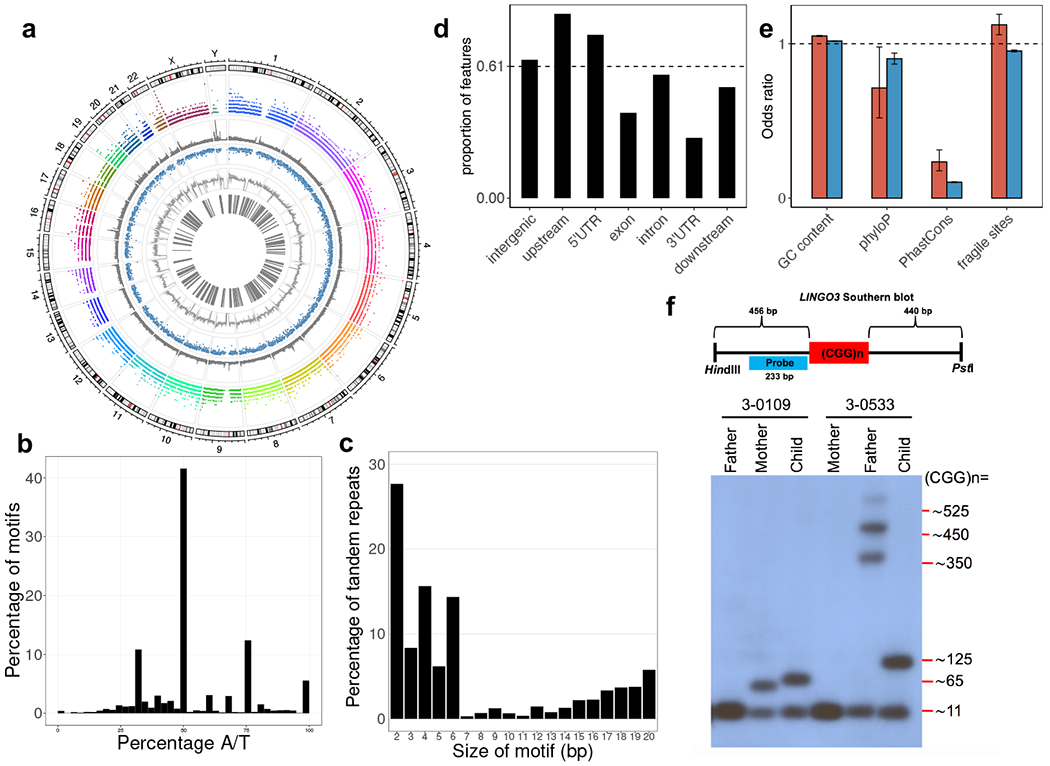
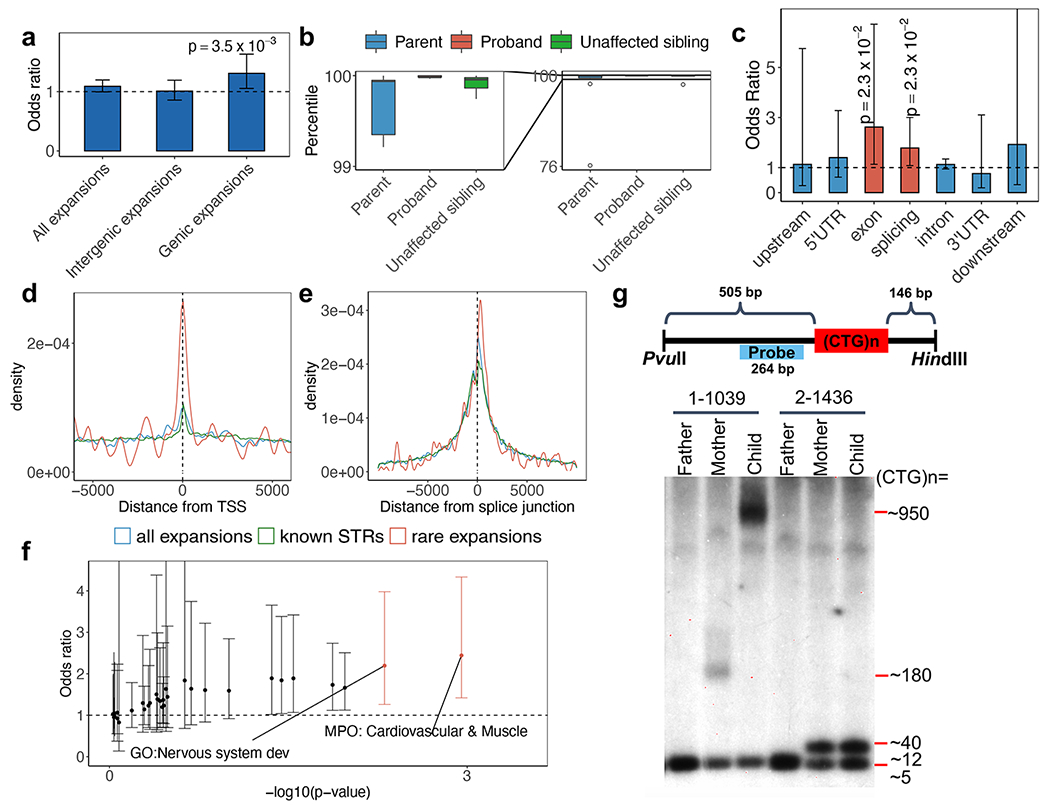
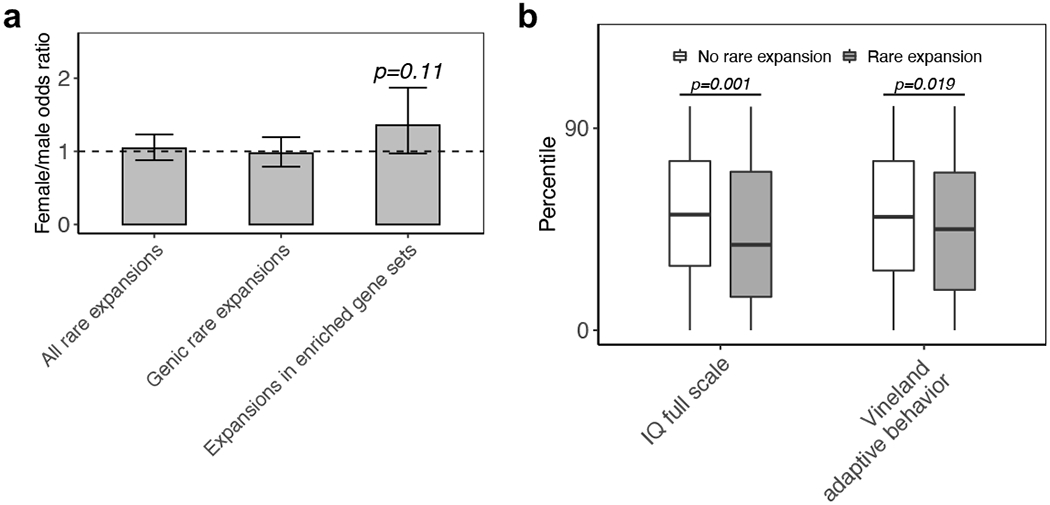
Comment in
-
Repeat DNA expands our understanding of autism spectrum disorder.Nature. 2021 Jan;589(7841):200-202. doi: 10.1038/d41586-020-03658-7. Nature. 2021. PMID: 33442037 No abstract available.
References
-
- López Castel A, Cleary JD & Pearson CE Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11, 165–70 (2010). - PubMed
-
- Fischbach GD & Lord C The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–5 (2010). - PubMed
References for Methods
-
- Ester M, Kriegel H, Sander J & Xu X A density-based algorithm for discovering clusters a density-based algorithm for discovering clusters in large spatial databases with noise. in Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (AAAI Press, 1996).
