

Polymorphic mobile element insertions contribute to gene expression and alternative splicing in human tissues
- PMID: 32718348
- PMCID: PMC7385971
- DOI: 10.1186/s13059-020-02101-4
Polymorphic mobile element insertions contribute to gene expression and alternative splicing in human tissues
Abstract
Background: Mobile elements are a major source of structural variants in the human genome, and some mobile elements can regulate gene expression and transcript splicing. However, the impact of polymorphic mobile element insertions (pMEIs) on gene expression and splicing in diverse human tissues has not been thoroughly studied. The multi-tissue gene expression and whole genome sequencing data generated by the Genotype-Tissue Expression (GTEx) project provide a great opportunity to systematically evaluate the role of pMEIs in regulating gene expression in human tissues.
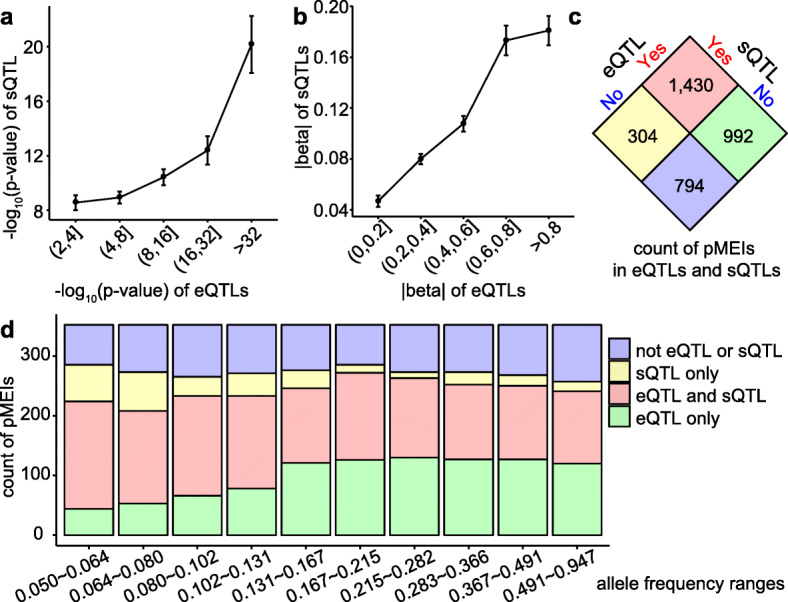
Results: Using the GTEx whole genome sequencing data, we identify 20,545 high-quality pMEIs from 639 individuals. Coupling pMEI genotypes with gene expression profiles, we identify pMEI-associated expression quantitative trait loci (eQTLs) and splicing quantitative trait loci (sQTLs) in 48 tissues. Using joint analyses of pMEIs and other genomic variants, pMEIs are predicted to be the potential causal variant for 3522 eQTLs and 3717 sQTLs. The pMEI-associated eQTLs and sQTLs show a high level of tissue specificity, and these pMEIs are enriched in the proximity of affected genes and in regulatory elements. Using reporter assays, we confirm that several pMEIs associated with eQTLs and sQTLs can alter gene expression levels and isoform proportions, respectively.
Conclusion: Overall, our study shows that pMEIs are associated with thousands of gene expression and splicing variations, indicating that pMEIs could have a significant role in regulating tissue-specific gene expression and transcript splicing. Detailed mechanisms for the role of pMEIs in gene regulation in different tissues will be an important direction for future studies.
Keywords: Alternative splicing; Gene expression regulation; Polymorphic mobile element insertions; Quantitative trait loci; Transposable elements.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
