

Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling
- PMID: 32725128
- PMCID: PMC7596811
- DOI: 10.1084/jem.20200600
Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling
Abstract
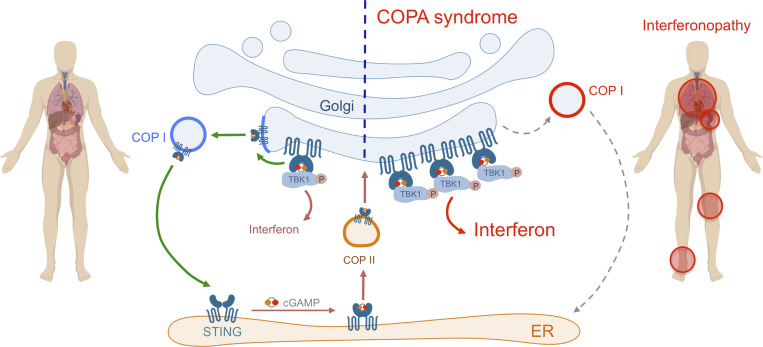
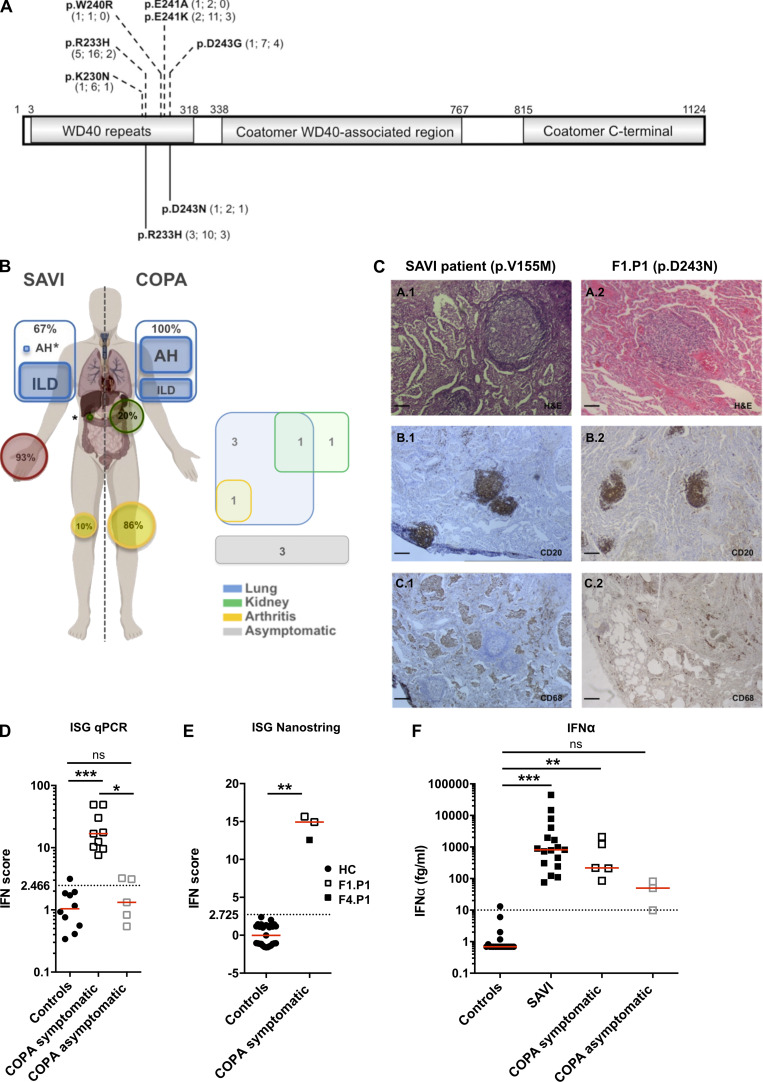
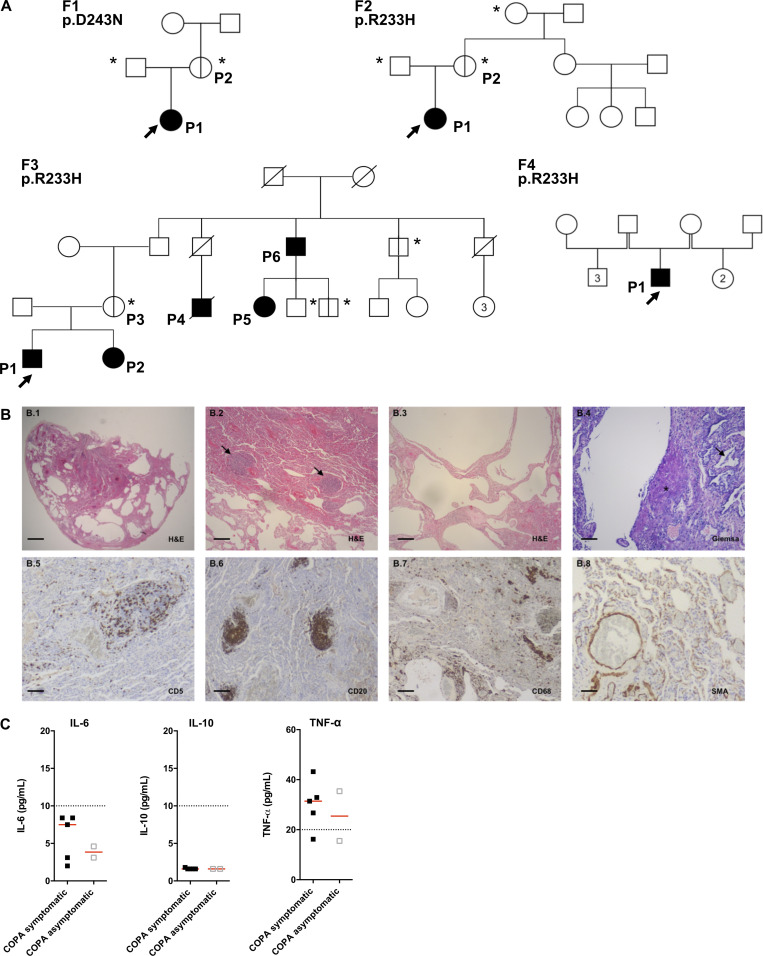
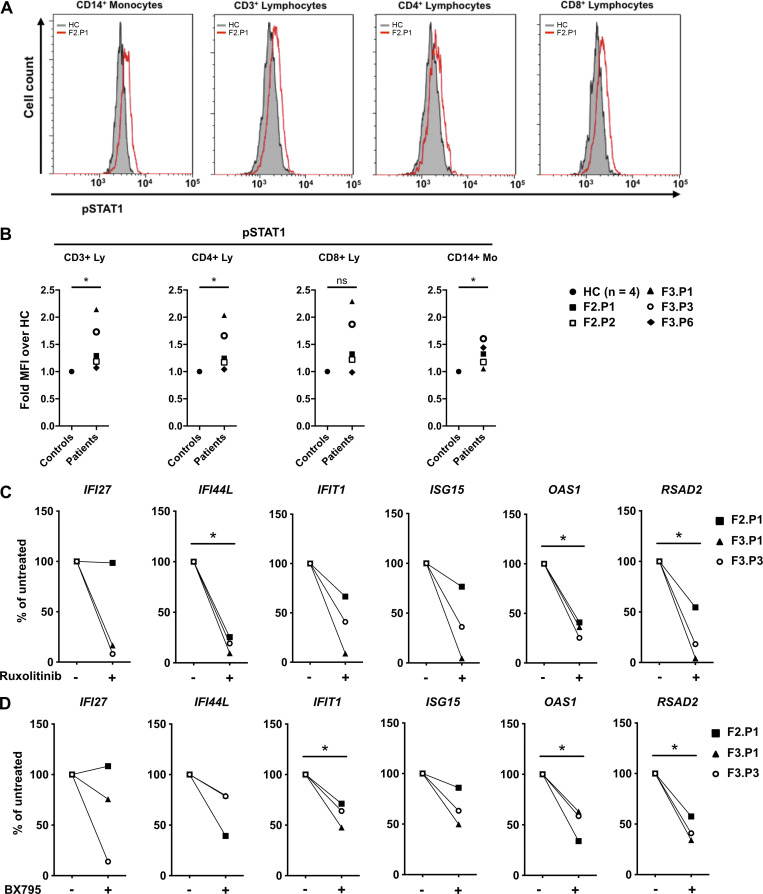
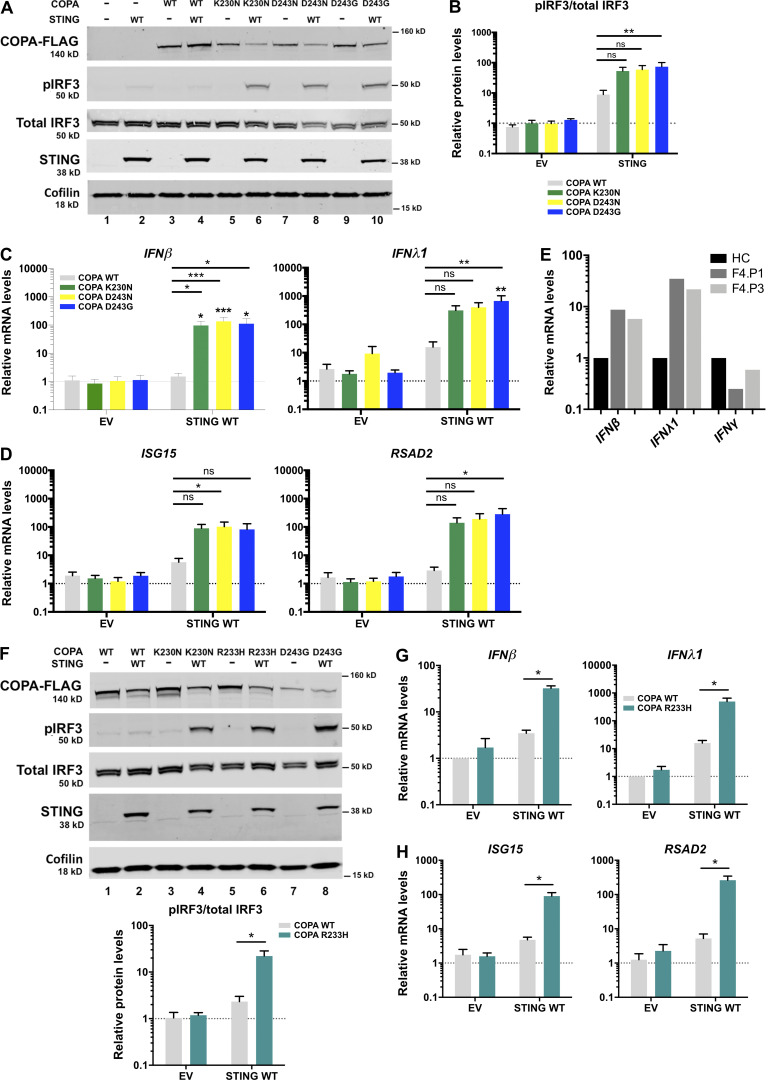
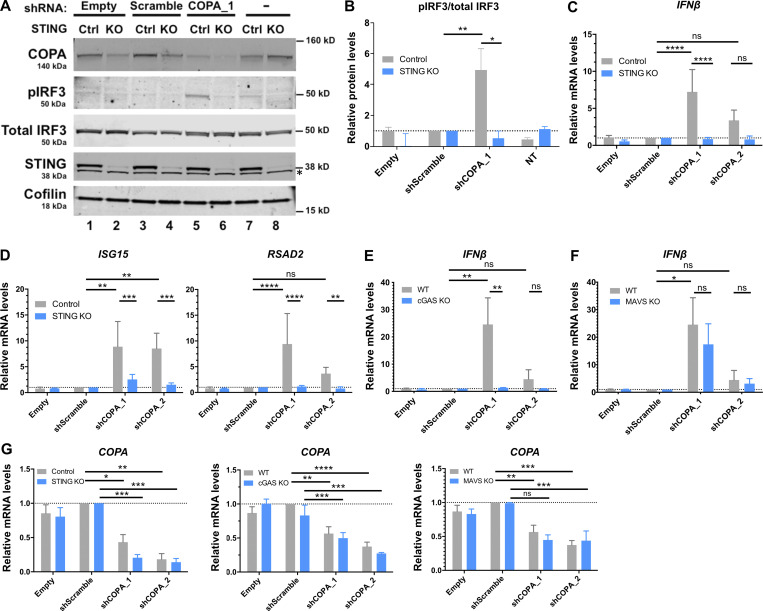
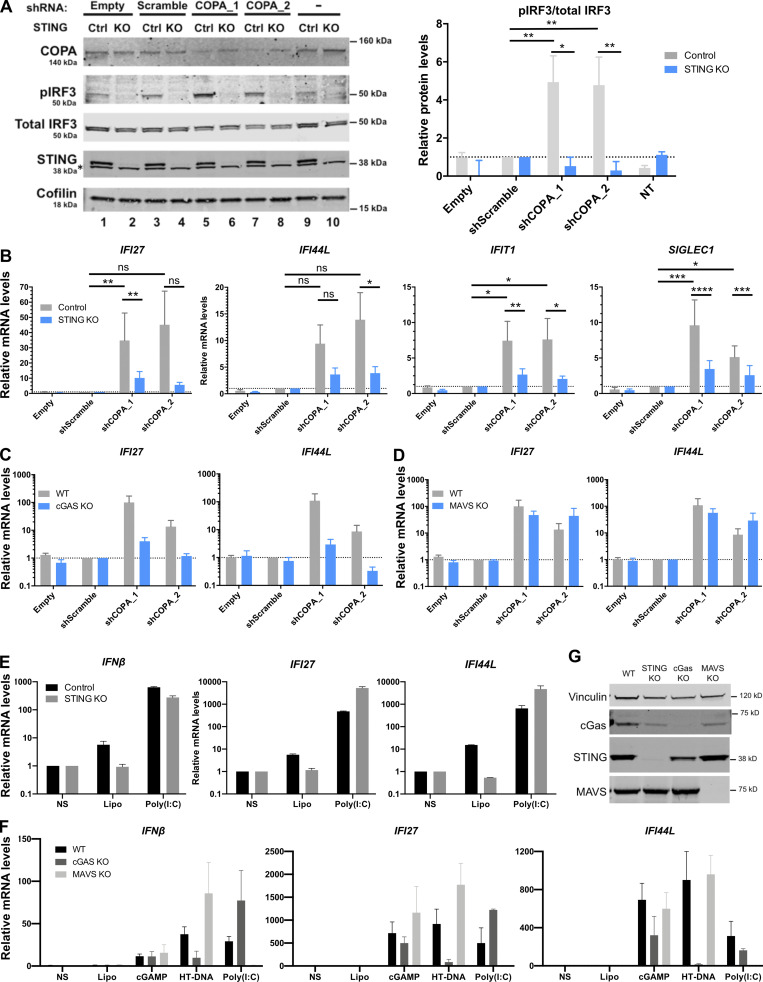
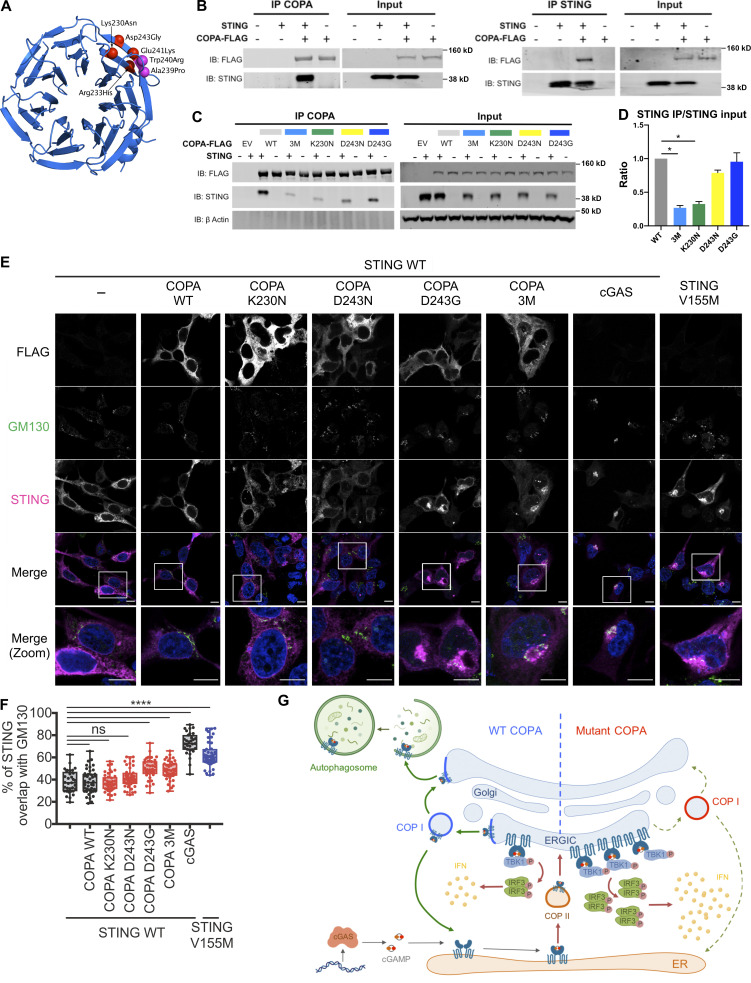
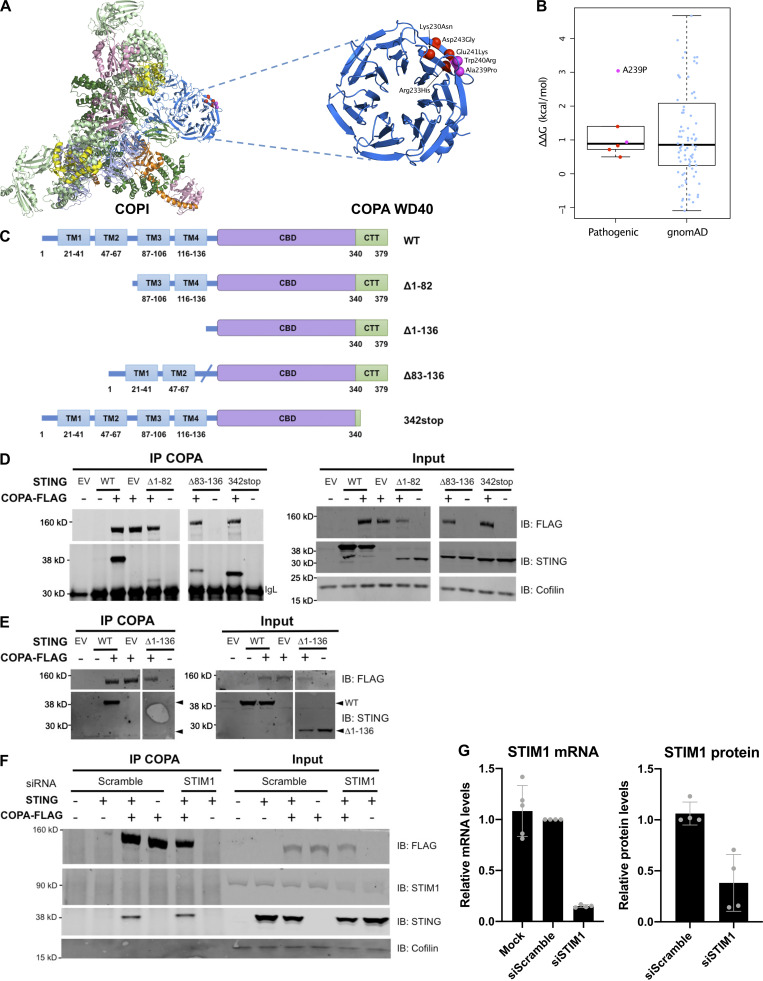
Heterozygous missense mutations in coatomer protein subunit α, COPA, cause a syndrome overlapping clinically with type I IFN-mediated disease due to gain-of-function in STING, a key adaptor of IFN signaling. Recently, increased levels of IFN-stimulated genes (ISGs) were described in COPA syndrome. However, the link between COPA mutations and IFN signaling is unknown. We observed elevated levels of ISGs and IFN-α in blood of symptomatic COPA patients. In vitro, both overexpression of mutant COPA and silencing of COPA induced STING-dependent IFN signaling. We detected an interaction between COPA and STING, and mutant COPA was associated with an accumulation of ER-resident STING at the Golgi. Given the known role of the coatomer protein complex I, we speculate that loss of COPA function leads to enhanced type I IFN signaling due to a failure of Golgi-to-ER STING retrieval. These data highlight the importance of the ER-Golgi axis in the control of autoinflammation and inform therapeutic strategies in COPA syndrome.
© 2020 Lepelley et al.
Conflict of interest statement
Disclosures: M. Wislez reported personal fees from Boeringher Ingelheim, Roche, MSD, BMS, Astra Zeneca, and Amgen outside the submitted work. Y.J. Crow reported "other" from Biogen outside the submitted work. No other disclosures were reported.
Figures
Comment in
-
COPA silences STING.J Exp Med. 2020 Nov 2;217(11):e20201517. doi: 10.1084/jem.20201517. J Exp Med. 2020. PMID: 32991673 Free PMC article.
References
-
- Briand C., Frémond M.-L., Bessis D., Carbasse A., Rice G.I., Bondet V., Duffy D., Chatenoud L., Blanche S., Crow Y.J., et al. . 2019. Efficacy of JAK1/2 inhibition in the treatment of chilblain lupus due to TREX1 deficiency. Ann. Rheum. Dis. 78:431–433. 10.1136/annrheumdis-2018-214037 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
