

Syntheses of Thailandepsin B Pseudo-Natural Products: Access to New Highly Potent HDAC Inhibitors via Late-Stage Modification
- PMID: 32725698
- PMCID: PMC7756392
- DOI: 10.1002/chem.202002449
Syntheses of Thailandepsin B Pseudo-Natural Products: Access to New Highly Potent HDAC Inhibitors via Late-Stage Modification
Abstract
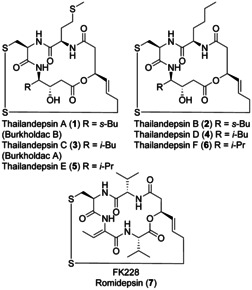
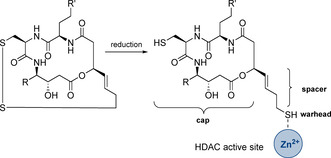
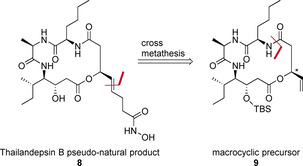
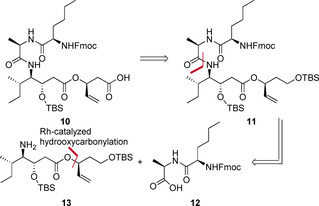
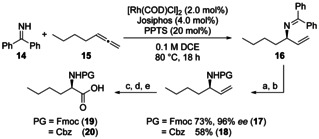
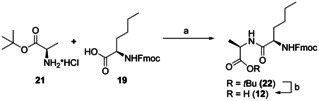
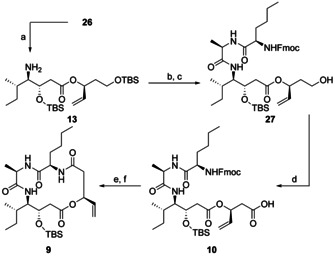
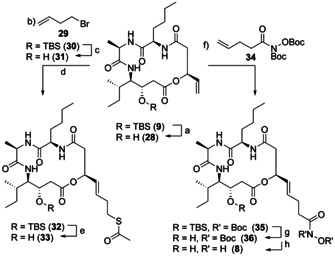
New Thailandepsin B pseudo-natural products have been prepared. Our synthetic strategy offers the possibility to introduce varying warheads via late stage modification. Additionally, it gives access to the asymmetric branched allylic ester moiety of the natural product in a highly diastereoselective manner applying rhodium-catalyzed hydrooxycarbonylation. The newly developed pseudo-natural products are extremely potent and selective HDAC inhibitors. The non-proteinogenic amino acid d-norleucine was obtained enantioselectively by a recently developed method of rhodium-catalyzed hydroamination.
Keywords: asymmetric catalysis; histone deacetylase inhibitors; rhodium; synthetic methods; thailandepsin.
© 2020 The Authors. Published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- None
-
- Cheng Y.-Q., Wang C., US Patent Application 20110060021, 2011.
Grants and funding
LinkOut - more resources
Full Text Sources
