

The ties that bind: functional clusters in limb-girdle muscular dystrophy
- PMID: 32727611
- PMCID: PMC7389686
- DOI: 10.1186/s13395-020-00240-7
The ties that bind: functional clusters in limb-girdle muscular dystrophy
Abstract
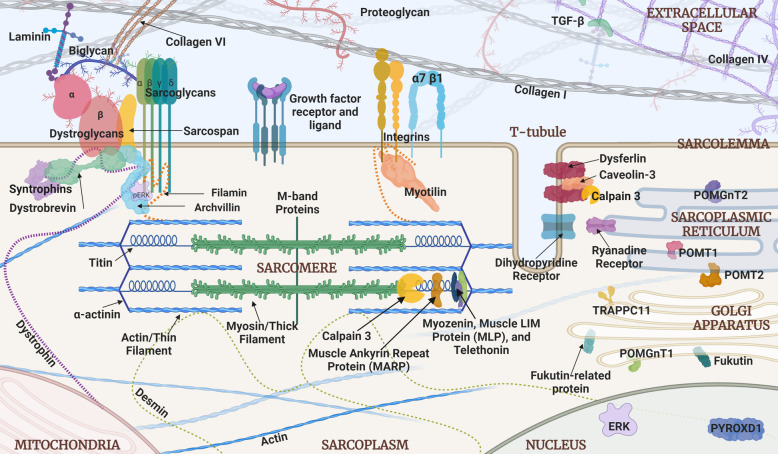
The limb-girdle muscular dystrophies (LGMDs) are a genetically pleiomorphic class of inherited muscle diseases that are known to share phenotypic features. Selected LGMD genetic subtypes have been studied extensively in affected humans and various animal models. In some cases, these investigations have led to human clinical trials of potential disease-modifying therapies, including gene replacement strategies for individual subtypes using adeno-associated virus (AAV) vectors. The cellular localizations of most proteins associated with LGMD have been determined. However, the functions of these proteins are less uniformly characterized, thus limiting our knowledge of potential common disease mechanisms across subtype boundaries. Correspondingly, broad therapeutic strategies that could each target multiple LGMD subtypes remain less developed. We believe that three major "functional clusters" of subcellular activities relevant to LGMD merit further investigation. The best known of these is the glycosylation modifications associated with the dystroglycan complex. The other two, mechanical signaling and mitochondrial dysfunction, have been studied less systematically but are just as promising with respect to the identification of significant mechanistic subgroups of LGMD. A deeper understanding of these disease pathways could yield a new generation of precision therapies that would each be expected to treat a broader range of LGMD patients than a single subtype, thus expanding the scope of the molecular medicines that may be developed for this complex array of muscular dystrophies.
Conflict of interest statement
PBK consults for AveXis, receives honoraria from Wiley for serving as associate editor of
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
