

15-Keto-PGE2 acts as a biased/partial agonist to terminate PGE2-evoked signaling
- PMID: 32727851
- PMCID: PMC7504929
- DOI: 10.1074/jbc.RA120.013988
15-Keto-PGE2 acts as a biased/partial agonist to terminate PGE2-evoked signaling
Abstract
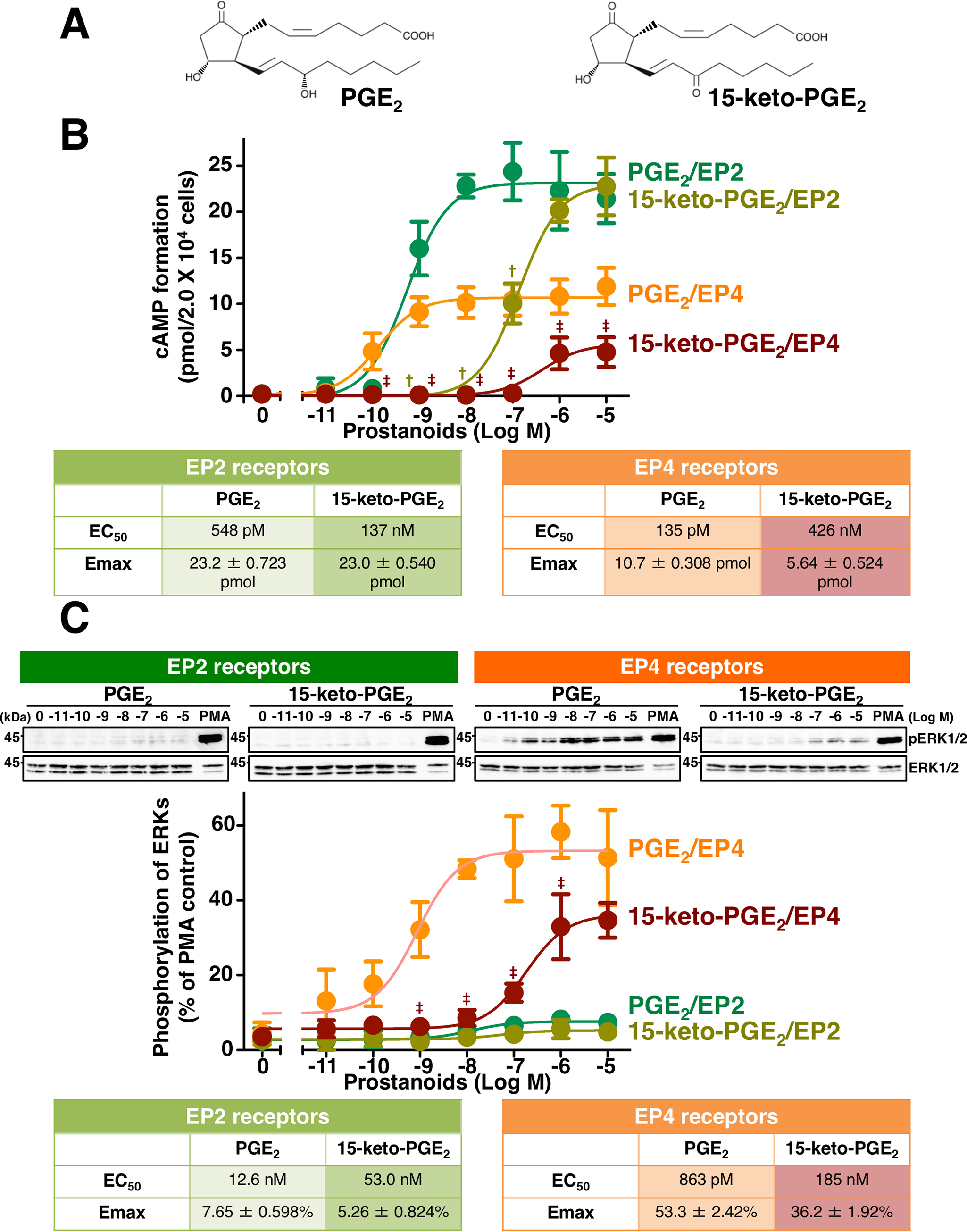
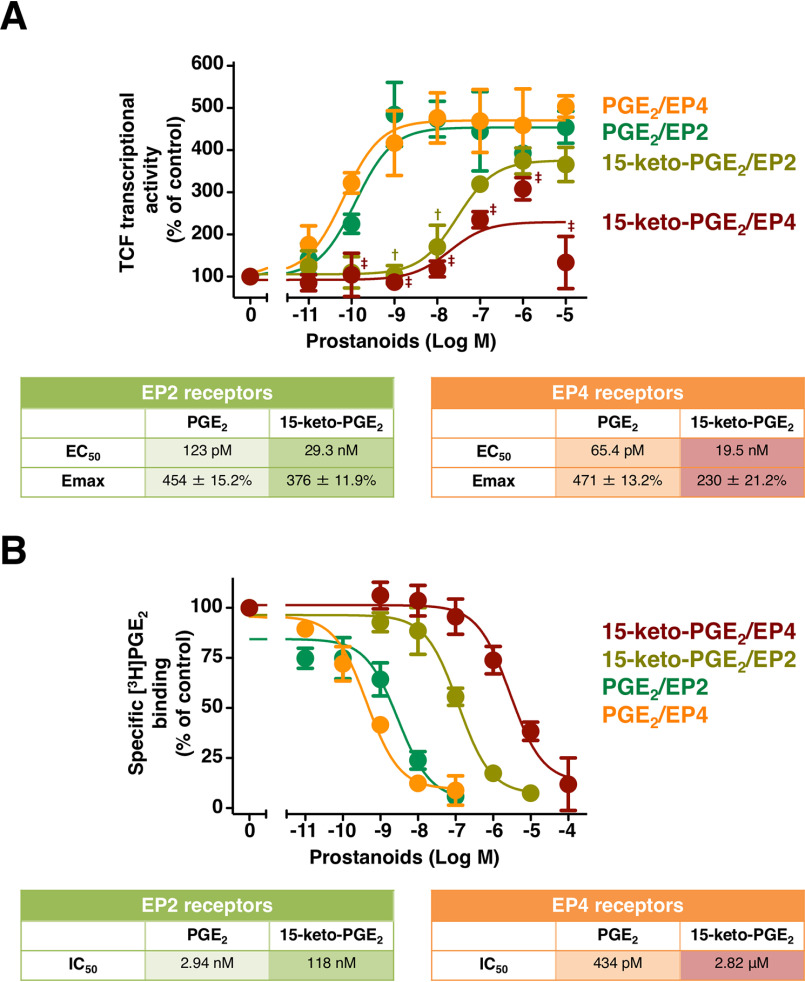
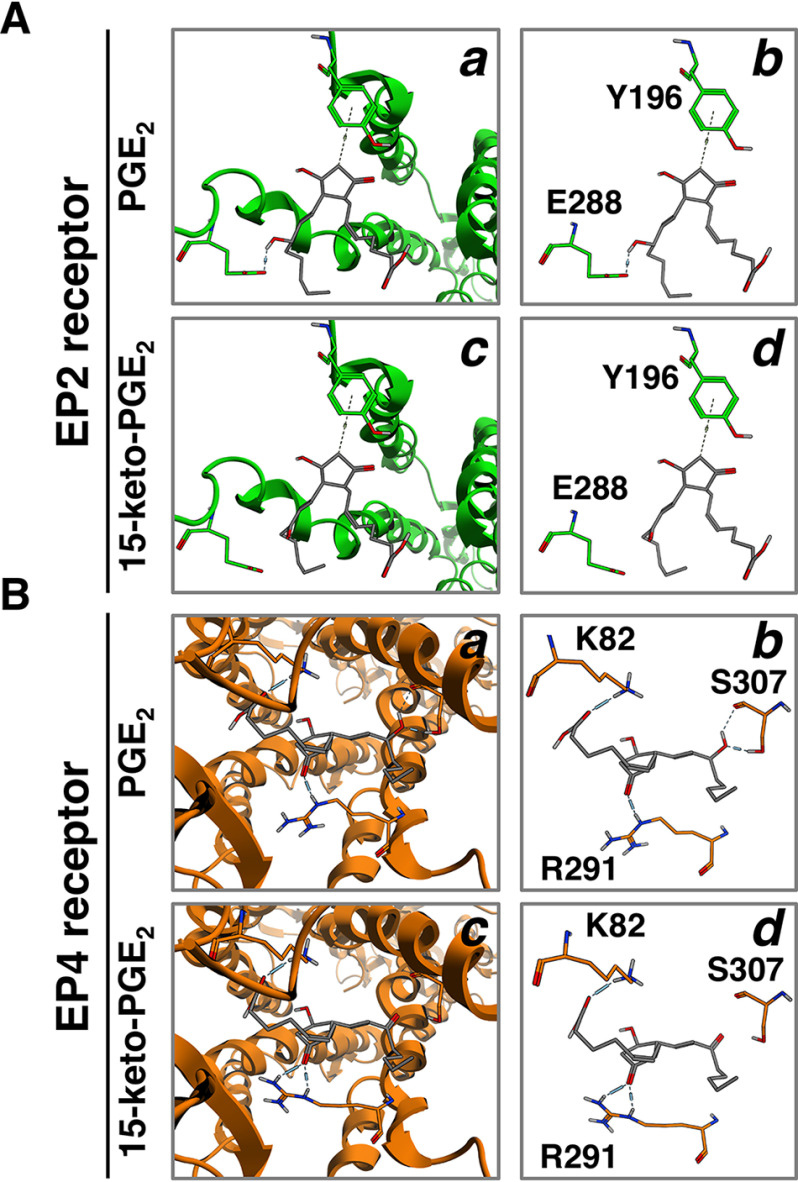
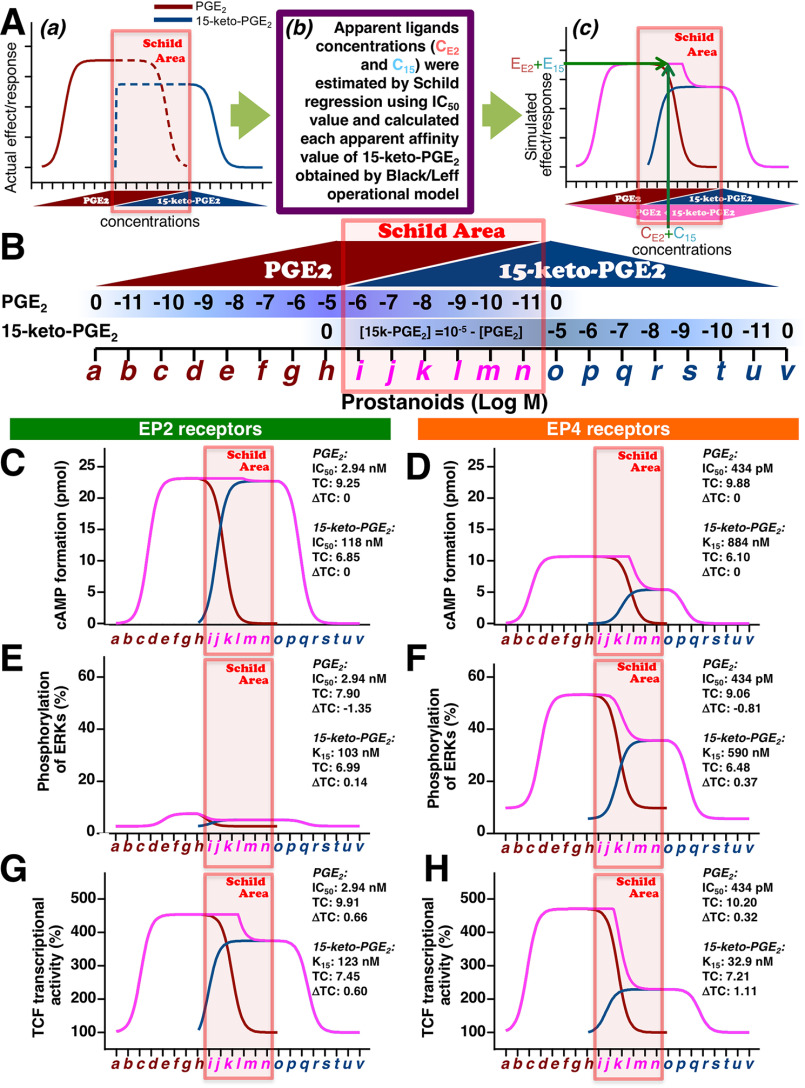
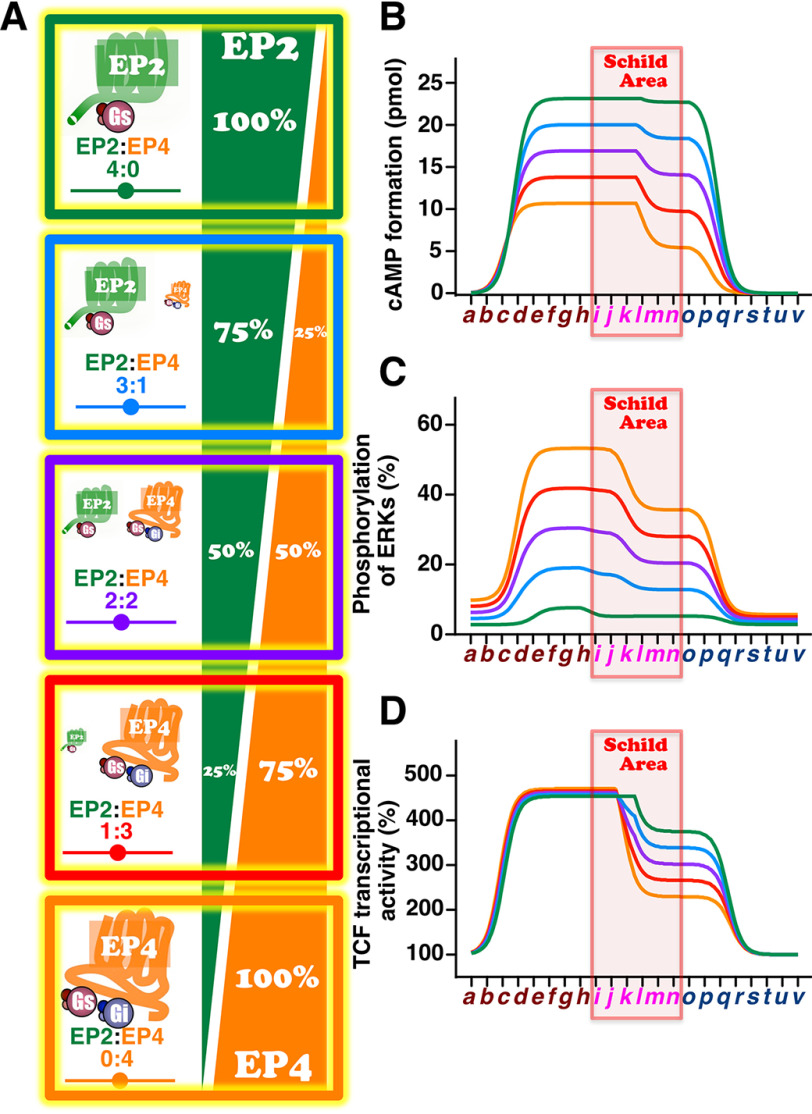
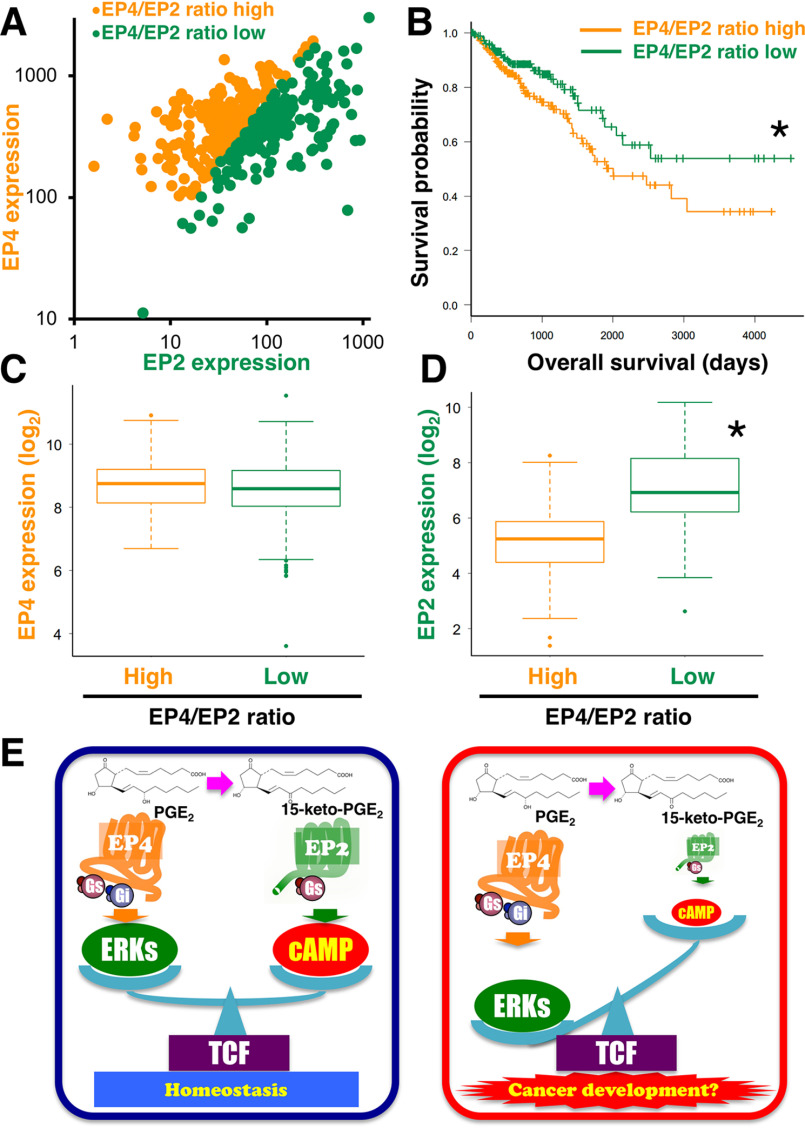
Prostaglandin E2 (PGE2) is well-known as an endogenous proinflammatory prostanoid synthesized from arachidonic acid by the activation of cyclooxygenase-2. E type prostanoid (EP) receptors are cognates for PGE2 that have four main subtypes: EP1 to EP4. Of these, the EP2 and EP4 prostanoid receptors have been shown to couple to Gαs-protein and can activate adenylyl cyclase to form cAMP. Studies suggest that EP4 receptors are involved in colorectal homeostasis and cancer development, but further work is needed to identify the roles of EP2 receptors in these functions. After sufficient inflammation has been evoked by PGE2, it is metabolized to 15-keto-PGE2 Thus, 15-keto-PGE2 has long been considered an inactive metabolite of PGE2 However, it may have an additional role as a biased and/or partial agonist capable of taking over the actions of PGE2 to gradually terminate reactions. Here, using cell-based experiments and in silico simulations, we show that PGE2-activated EP4 receptor-mediated signaling may evoke the primary initiating reaction of the cells, which would take over the 15-keto-PGE2-activated EP2 receptor-mediated signaling after PGE2 is metabolized to 15-keto-PGE2 The present results shed light on new aspects of 15-keto-PGE2, which may have important roles in passing on activities to EP2 receptors from PGE2-stimulated EP4 receptors as a "switched agonist." This novel mechanism may be significant for gradually terminating PGE2-evoked inflammation and/or maintaining homeostasis of colorectal tissues/cells functions.
Keywords: 15-keto-PGE2; EP2 prostanoid receptors; EP4 prostanoid receptors; G-protein–coupled receptor (GPCR); PGE2; Schild regression; biased agonist; biased ligand; bioinformatics; cancer biology; pharmacology; prostaglandin; switched agonist; switched ligand.
© 2020 Endo et al.
Conflict of interest statement
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Abramovitz M., Adam M., Boie Y., Carrière M., Denis D., Godbout C., Lamontagne S., Rochette C., Sawyer N., Tremblay N. M., Belley M., Gallant M., Dufresne C., Gareau Y., Ruel R., et al. (2000) The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analog. Biochim. Biophys. Acta 1483, 285–293 10.1016/S1388-1981(99)00164-X - DOI - PubMed
-
- Srivastava S., Verma M., and Henson D. E. (2001) Biomarkers for early detection of colon cancer. Clin. Cancer Res. 7, 1118–1126 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Research Materials
