

Epigenetic regulator function through mouse gastrulation
- PMID: 32728215
- PMCID: PMC7415732
- DOI: 10.1038/s41586-020-2552-x
Epigenetic regulator function through mouse gastrulation
Abstract
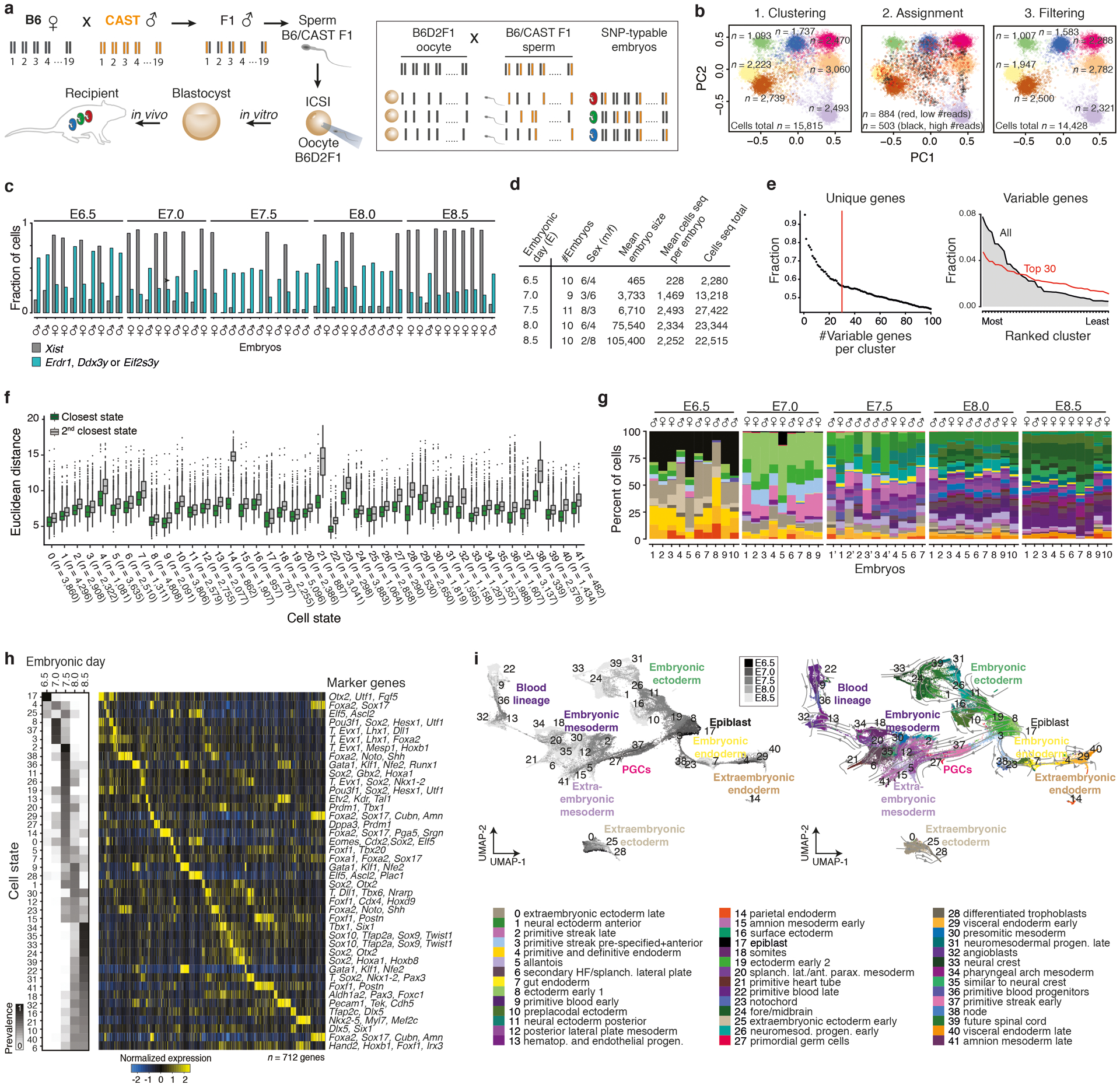
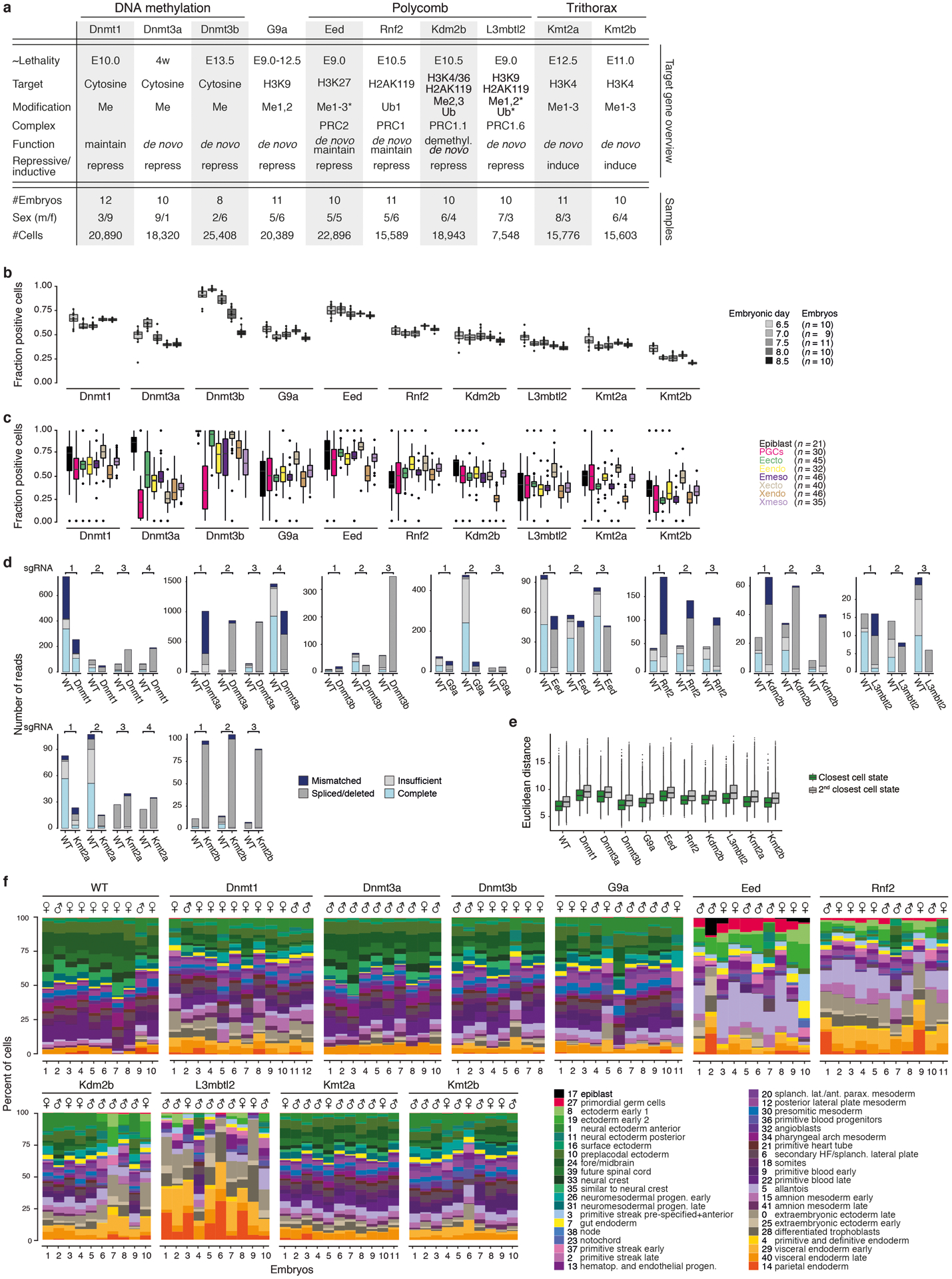
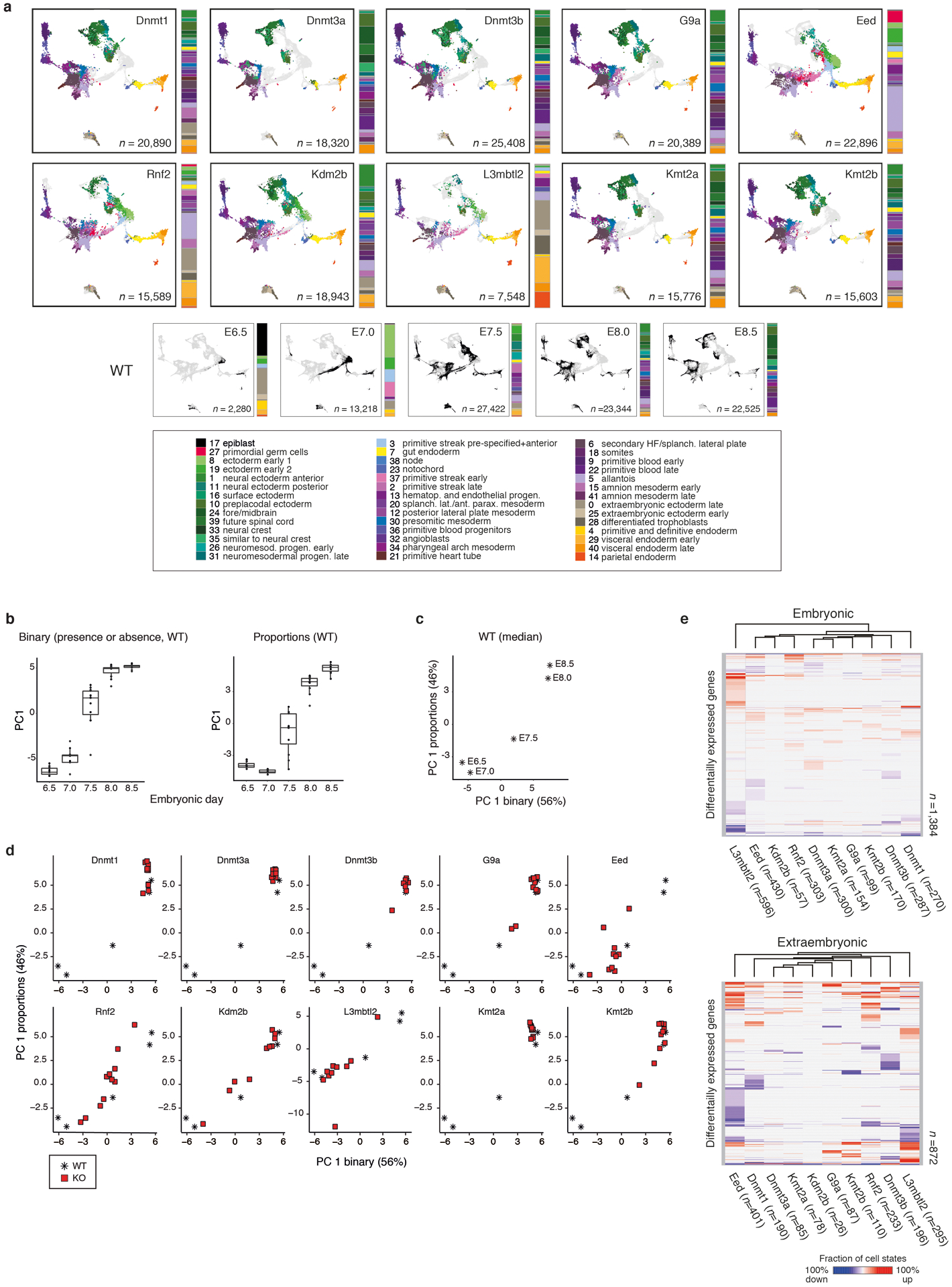
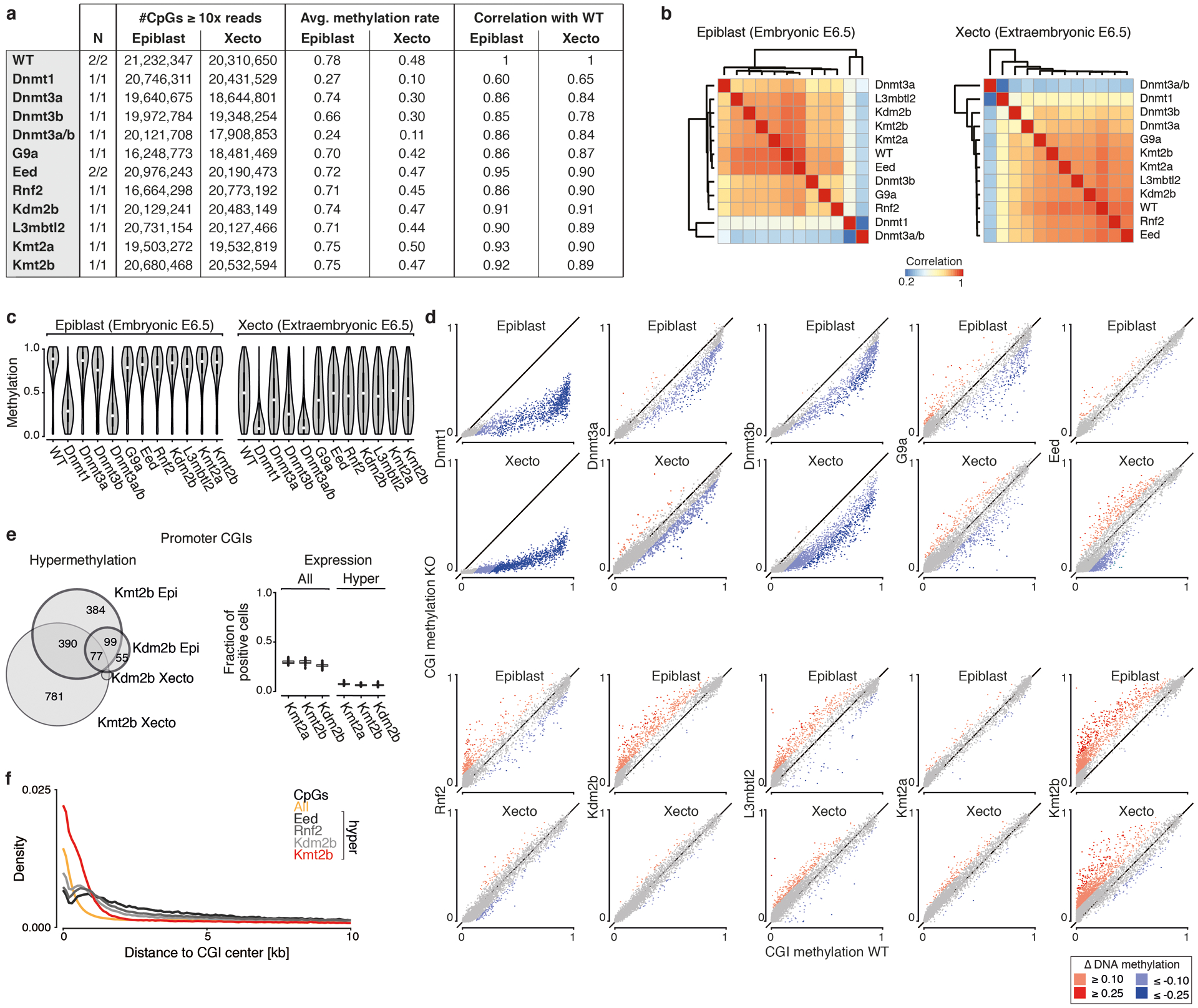
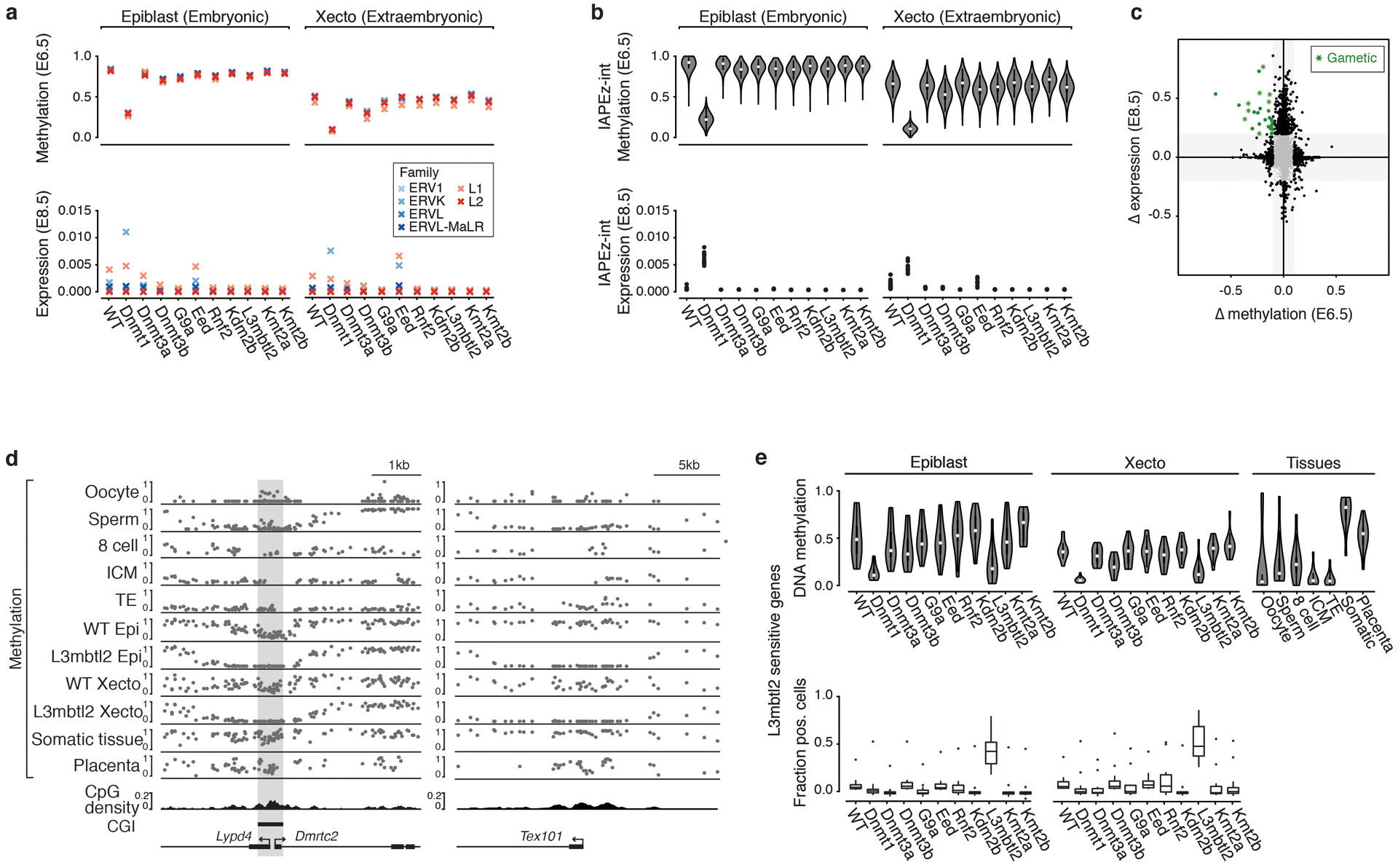
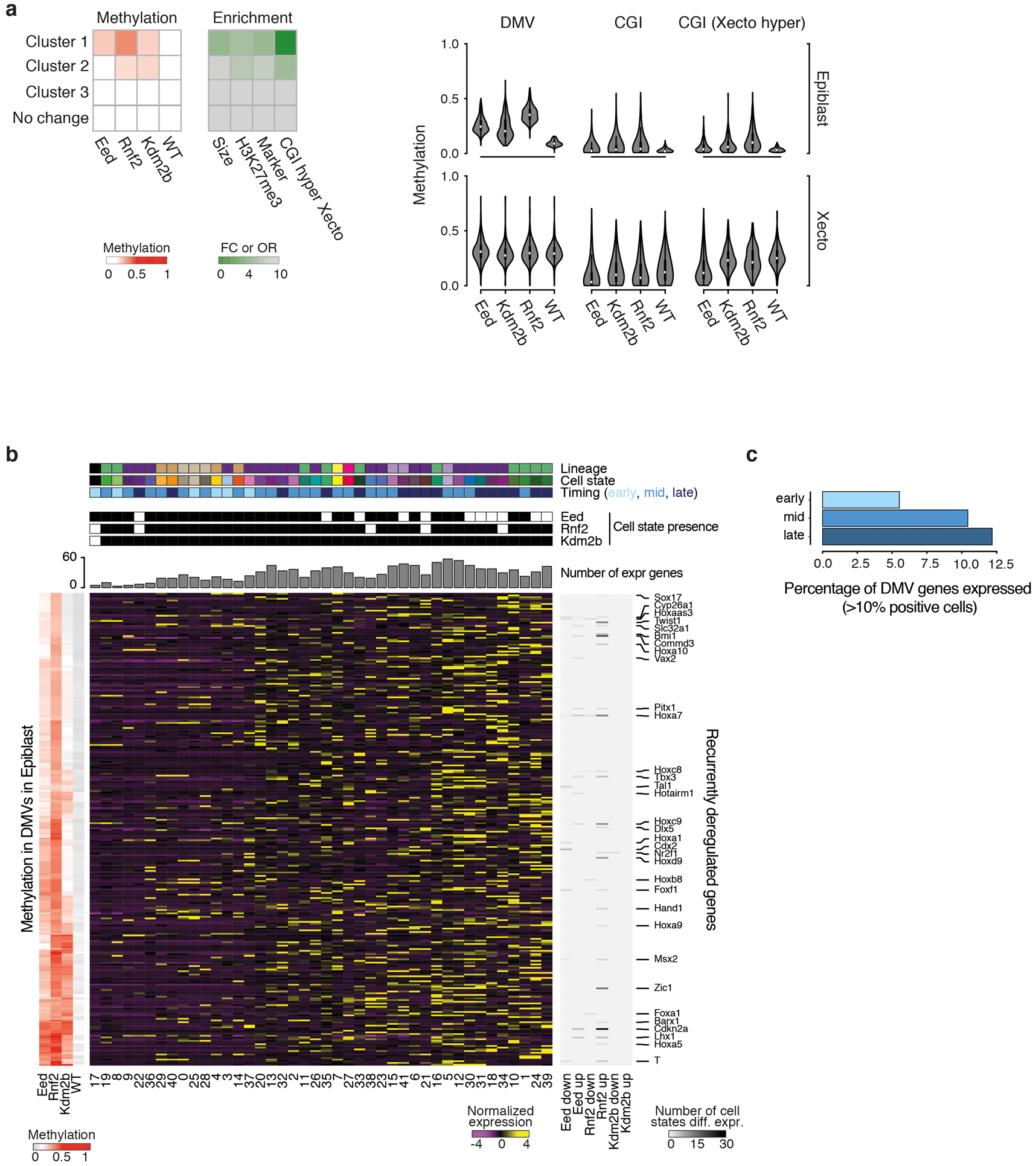
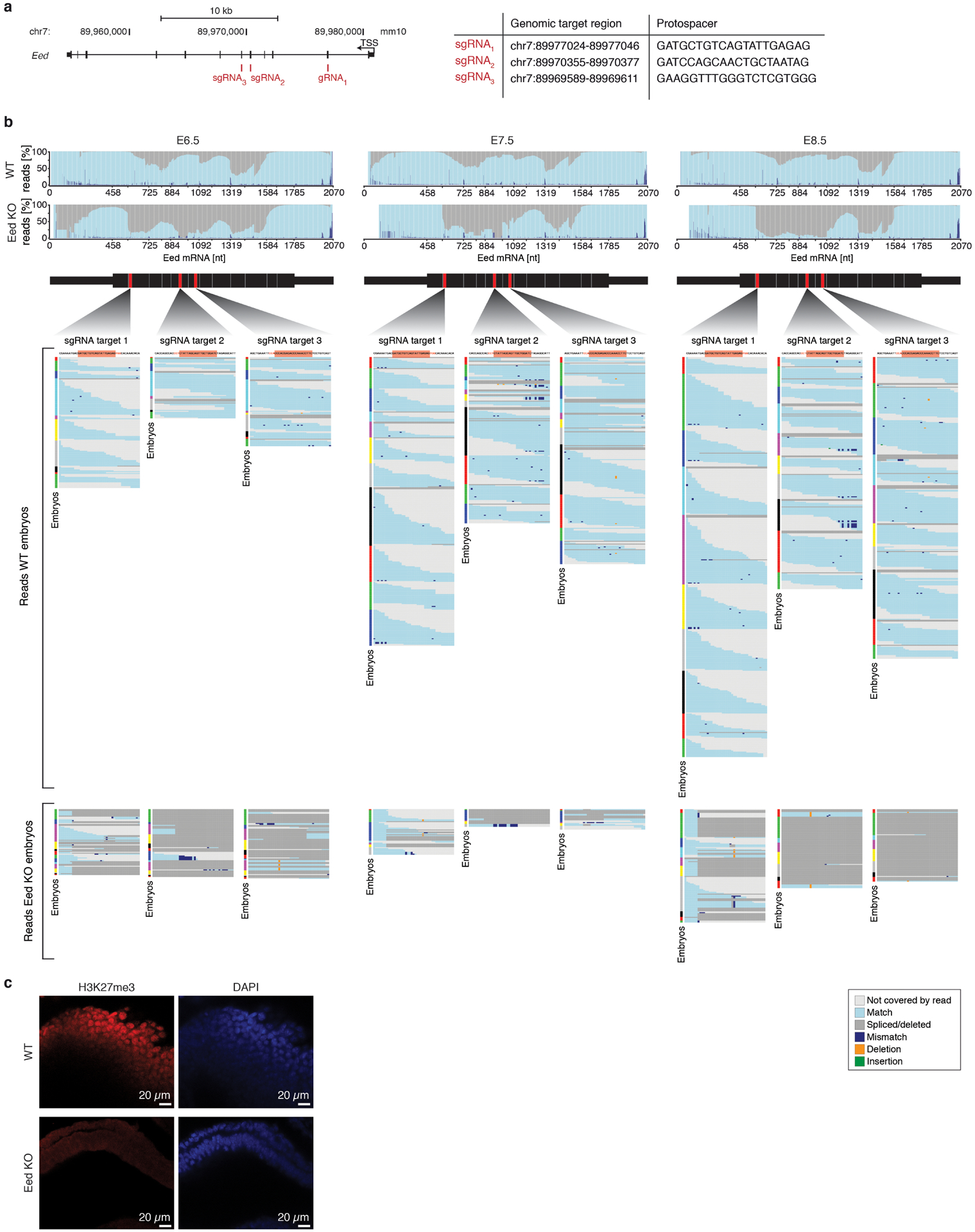
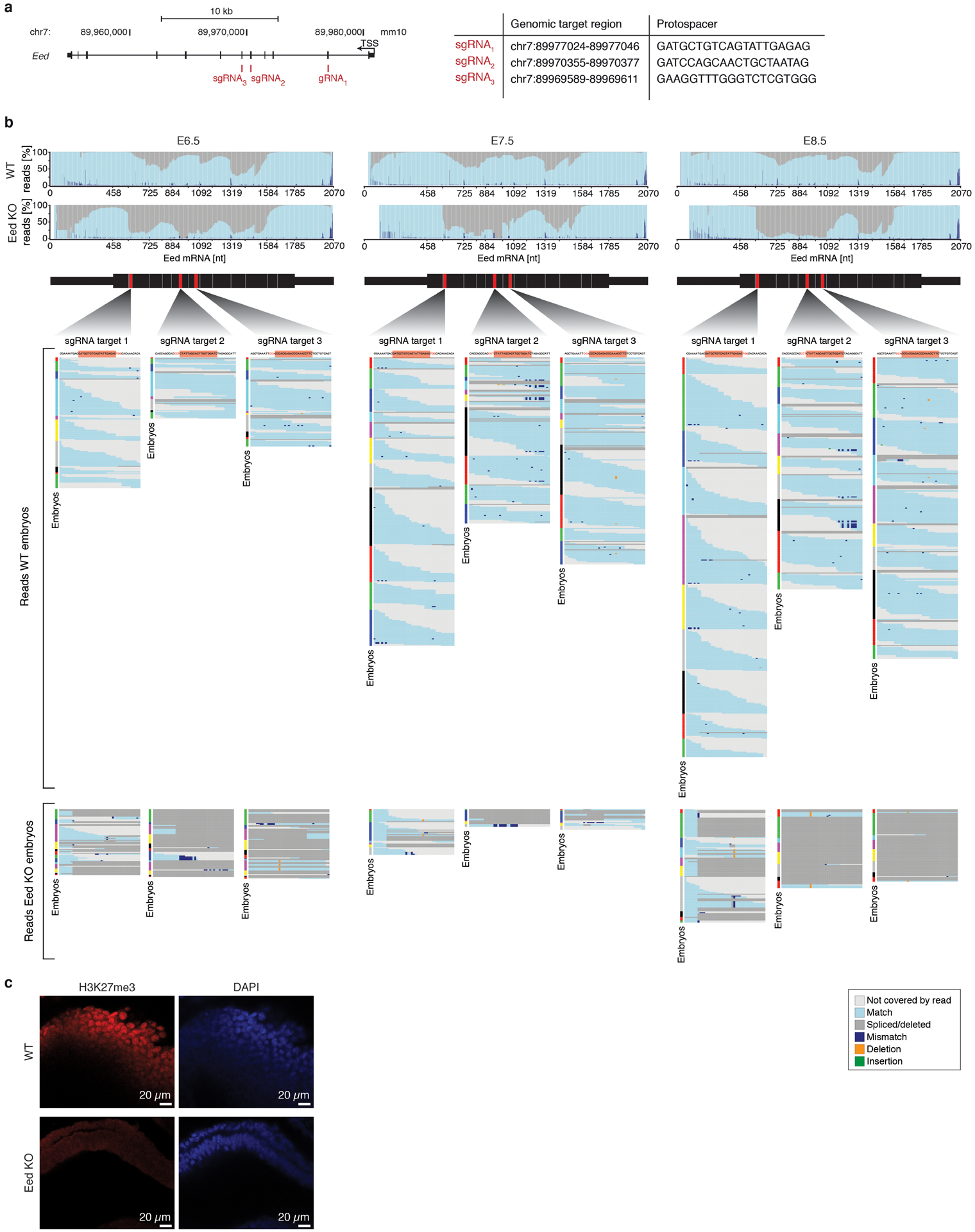
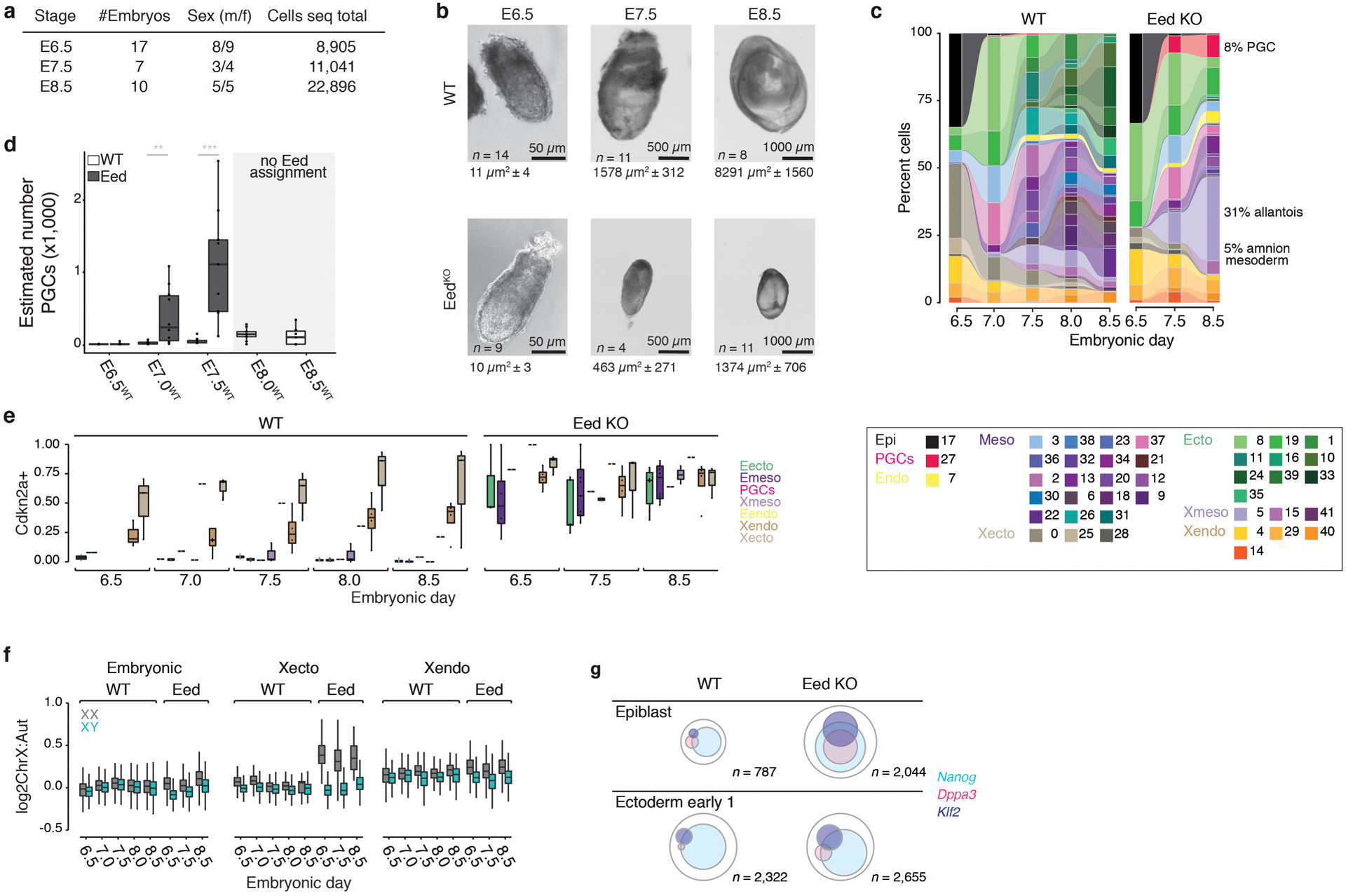
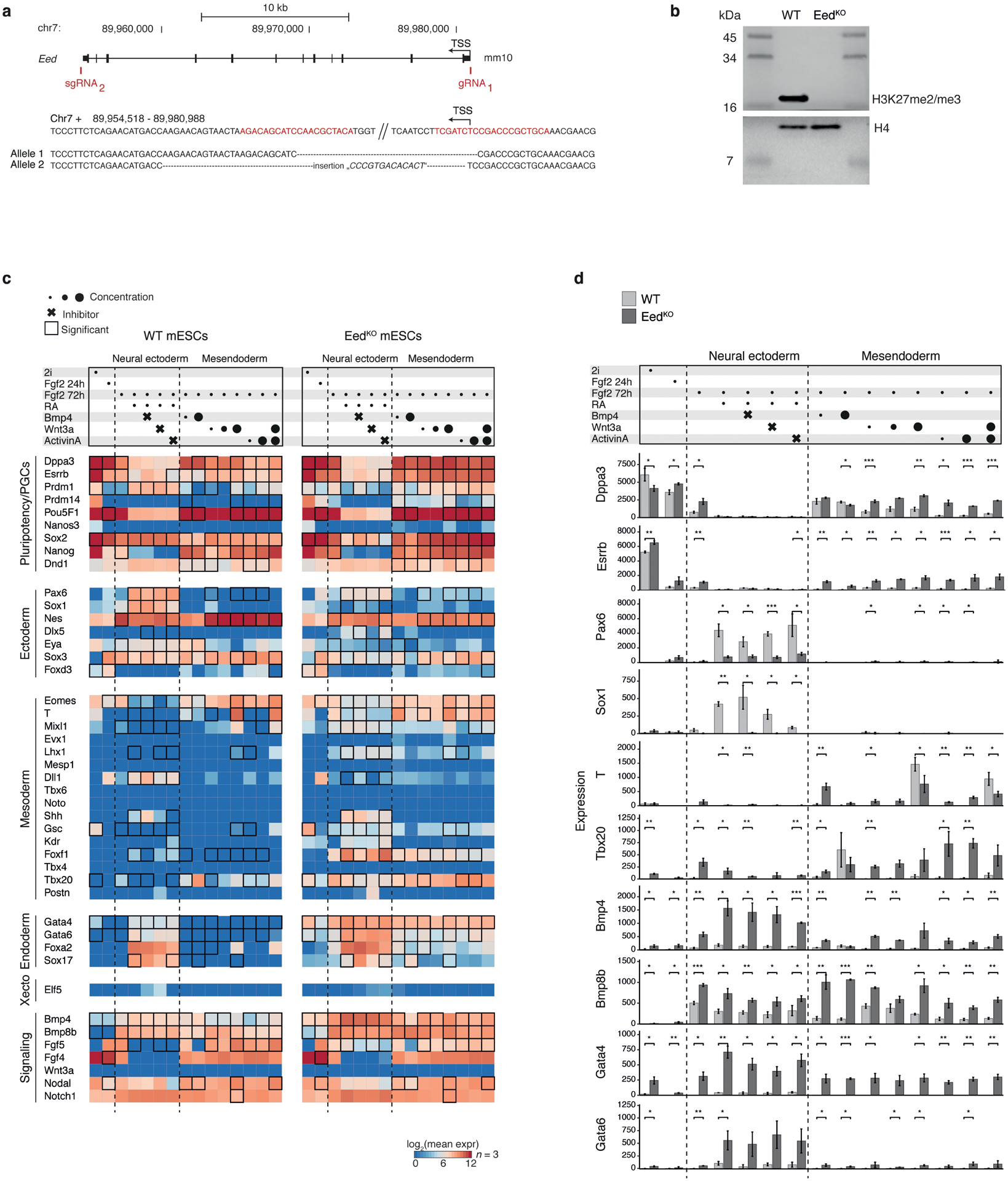
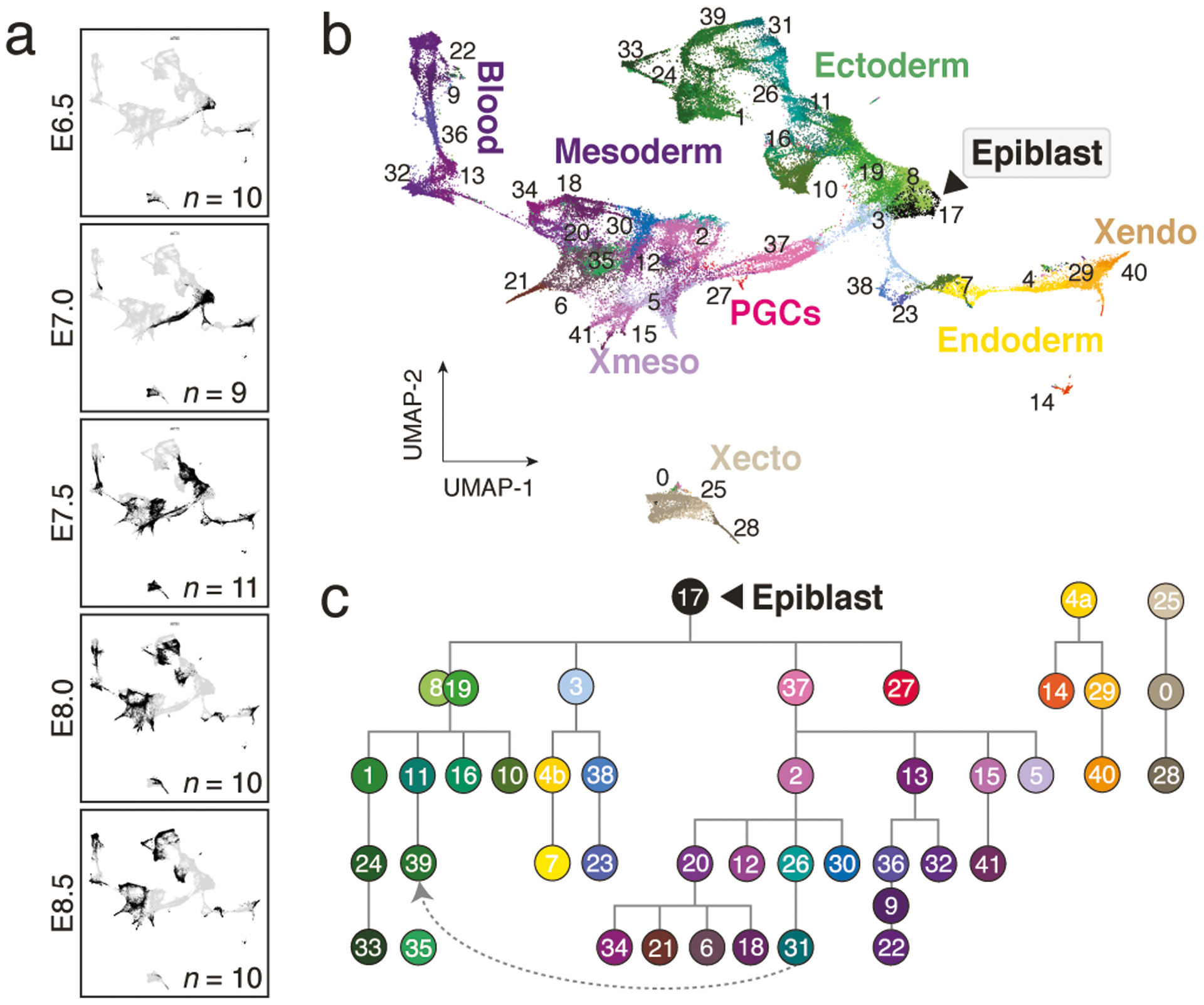
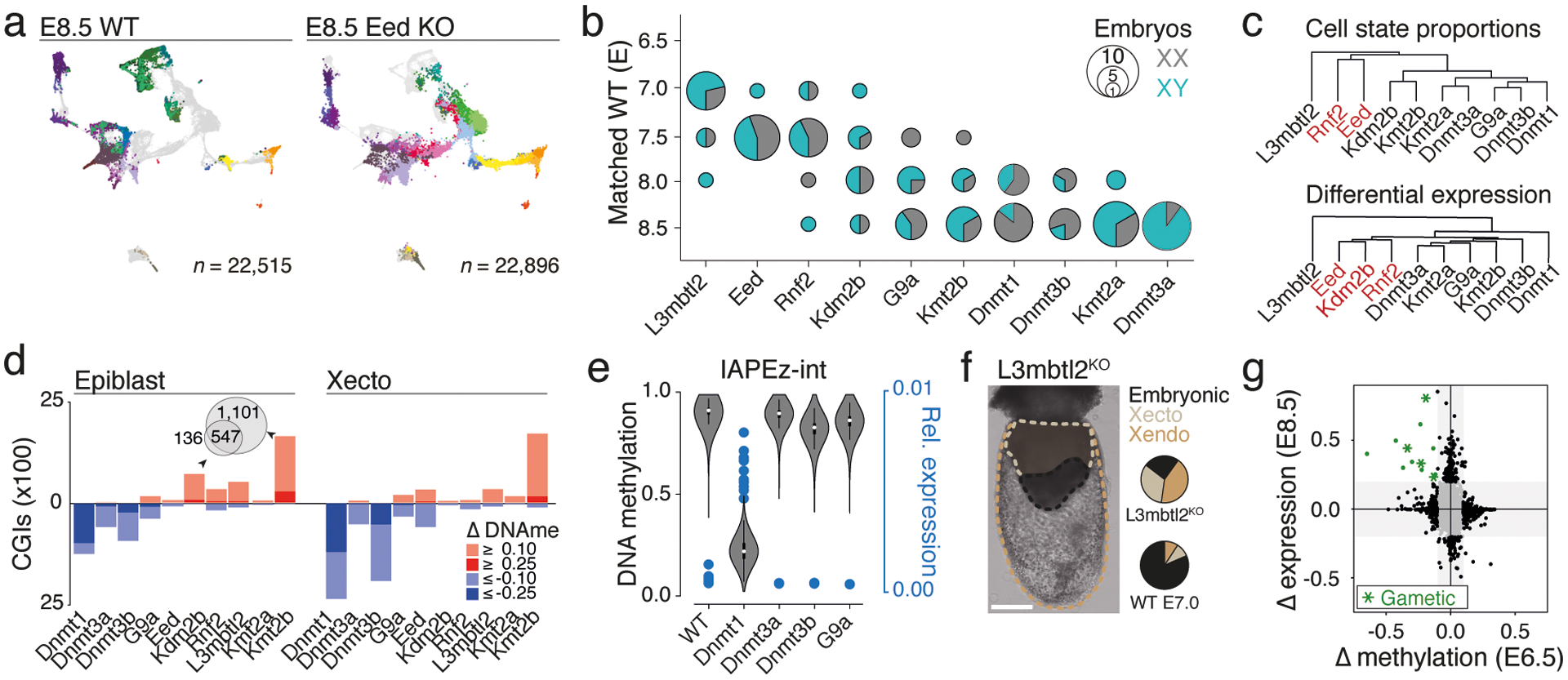
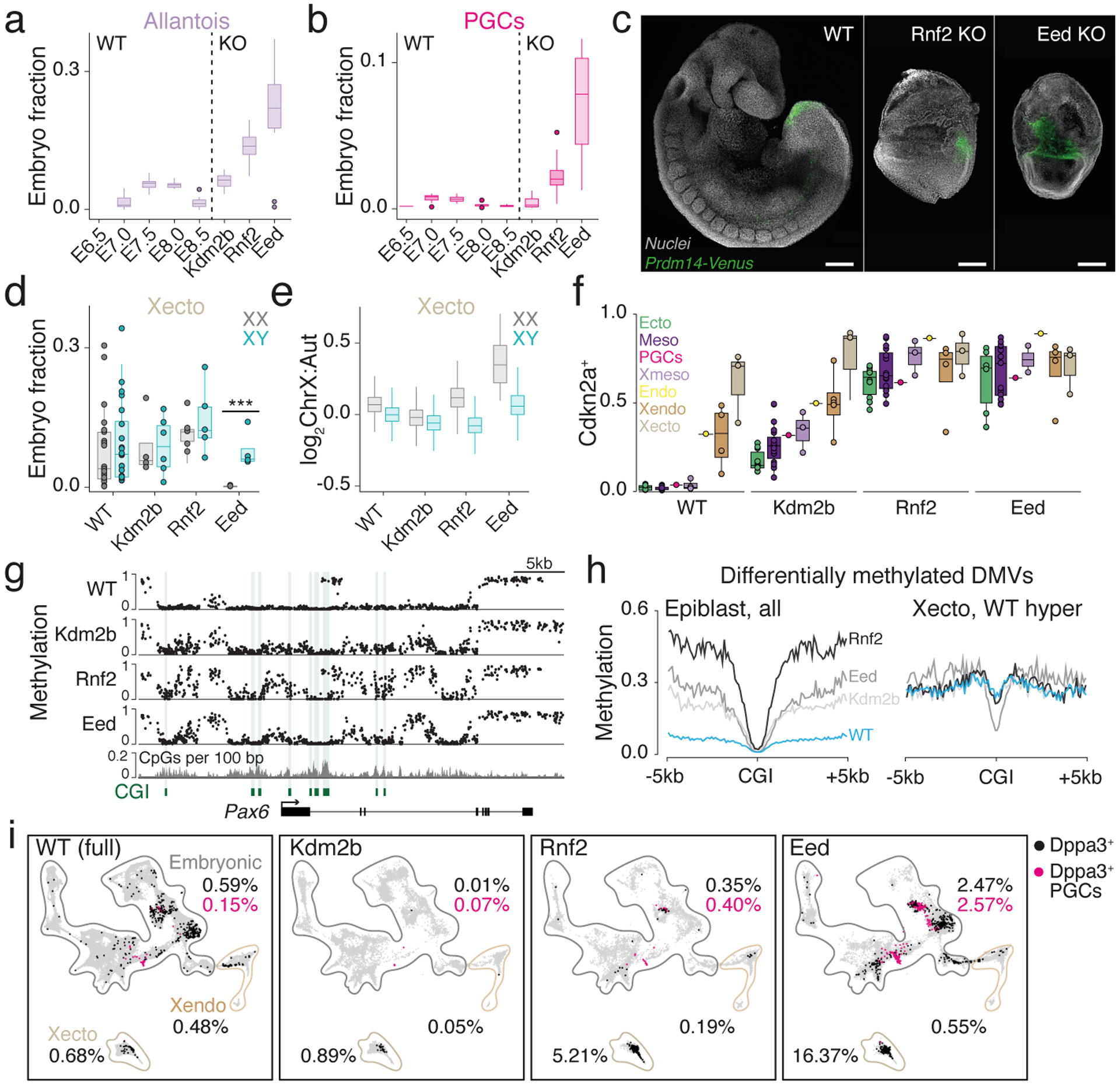
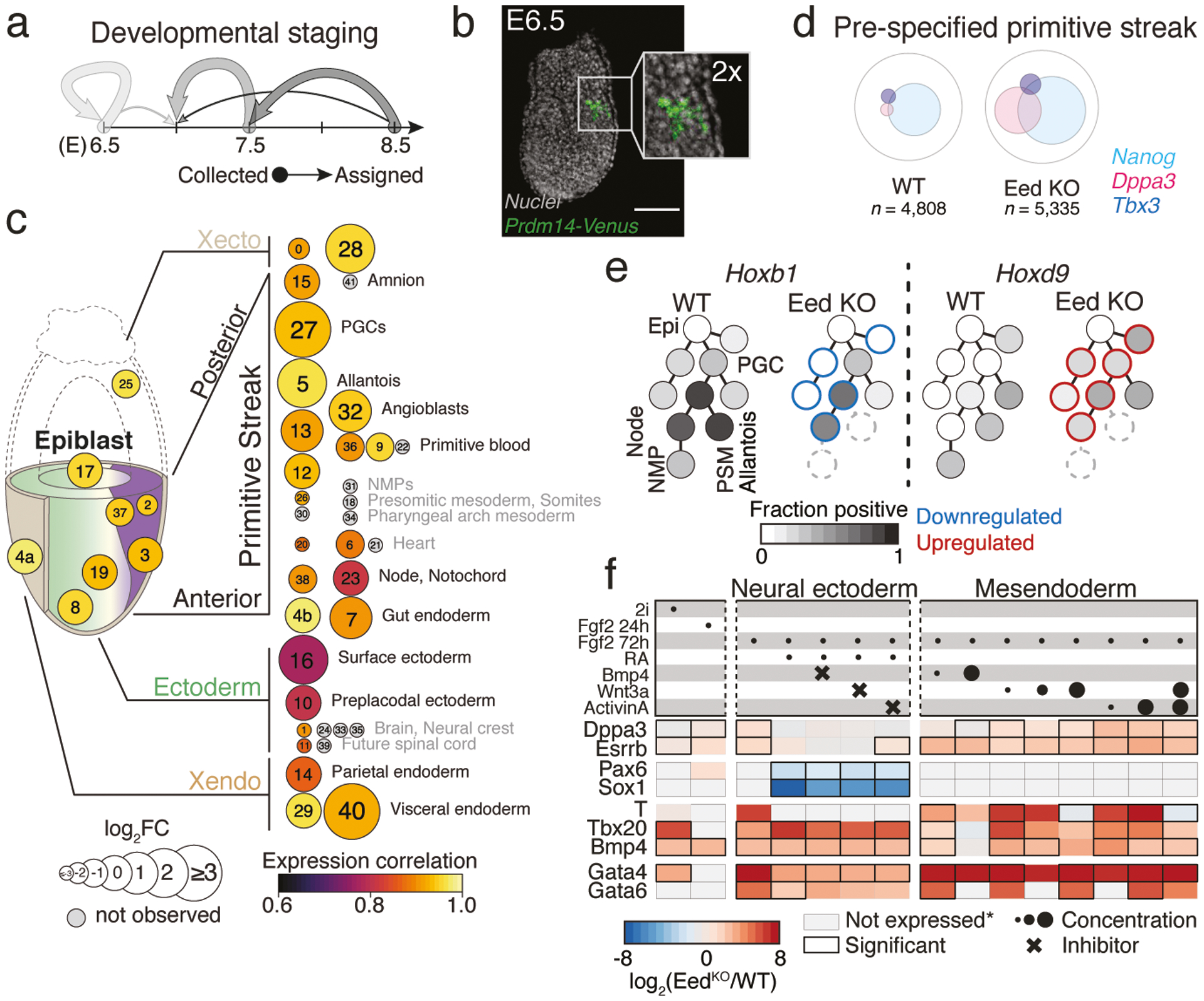
During ontogeny, proliferating cells become restricted in their fate through the combined action of cell-type-specific transcription factors and ubiquitous epigenetic machinery, which recognizes universally available histone residues or nucleotides in a context-dependent manner1,2. The molecular functions of these regulators are generally well understood, but assigning direct developmental roles to them is hampered by complex mutant phenotypes that often emerge after gastrulation3,4. Single-cell RNA sequencing and analytical approaches have explored this highly conserved, dynamic period across numerous model organisms5-8, including mouse9-18. Here we advance these strategies using a combined zygotic perturbation and single-cell RNA-sequencing platform in which many mutant mouse embryos can be assayed simultaneously, recovering robust morphological and transcriptional information across a panel of ten essential regulators. Deeper analysis of central Polycomb repressive complex (PRC) 1 and 2 components indicates substantial cooperativity, but distinguishes a dominant role for PRC2 in restricting the germline. Moreover, PRC mutant phenotypes emerge after gross epigenetic and transcriptional changes within the initial conceptus prior to gastrulation. Our experimental framework may eventually lead to a fully quantitative view of how cellular diversity emerges using an identical genetic template and from a single totipotent cell.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures

Comment in
-
Testing the developing epigenome.Nat Rev Genet. 2020 Oct;21(10):579. doi: 10.1038/s41576-020-00282-z. Nat Rev Genet. 2020. PMID: 32820272 No abstract available.
References
-
- Hemberger M, Dean W & Reik W Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nature Publishing Group 10, 526–537 (2009). - PubMed
-
- Meissner A Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol 28, 1079–1088 (2010). - PubMed
-
- Surani MA, Hayashi K & Hajkova P Genetic and epigenetic regulators of pluripotency. Cell 128, 747–762 (2007). - PubMed
-
- Plass M et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 360, eaaq1723 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
