

An atlas of dynamic chromatin landscapes in mouse fetal development
- PMID: 32728240
- PMCID: PMC7398618
- DOI: 10.1038/s41586-020-2093-3
An atlas of dynamic chromatin landscapes in mouse fetal development
Erratum in
-
Author Correction: An atlas of dynamic chromatin landscapes in mouse fetal development.Nature. 2020 Oct;586(7831):E31. doi: 10.1038/s41586-020-2841-4. Nature. 2020. PMID: 33037424 Free PMC article.
-
Author Correction: An atlas of dynamic chromatin landscapes in mouse fetal development.Nature. 2021 Jan;589(7842):E4. doi: 10.1038/s41586-020-03089-4. Nature. 2021. PMID: 33398137 Free PMC article. No abstract available.
Abstract
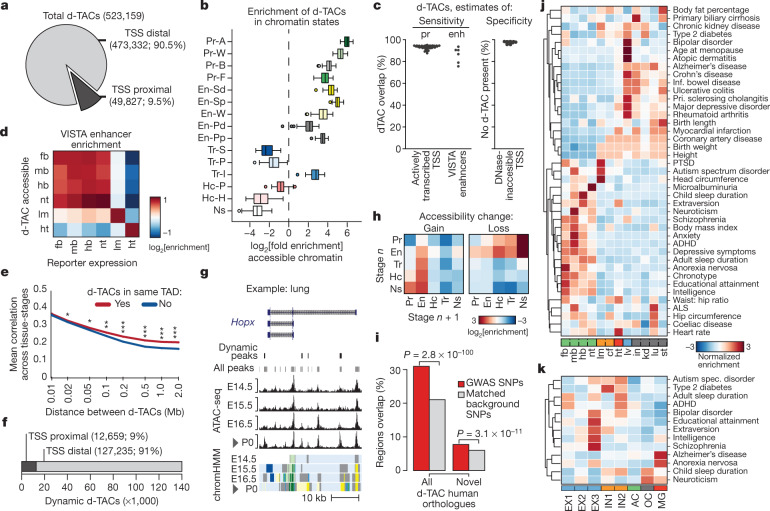
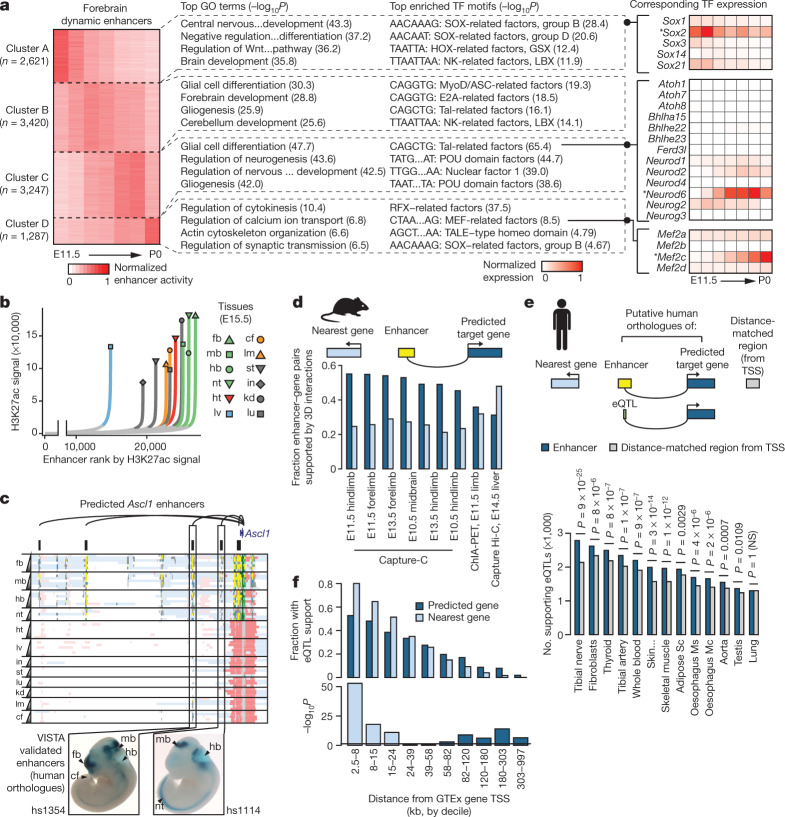
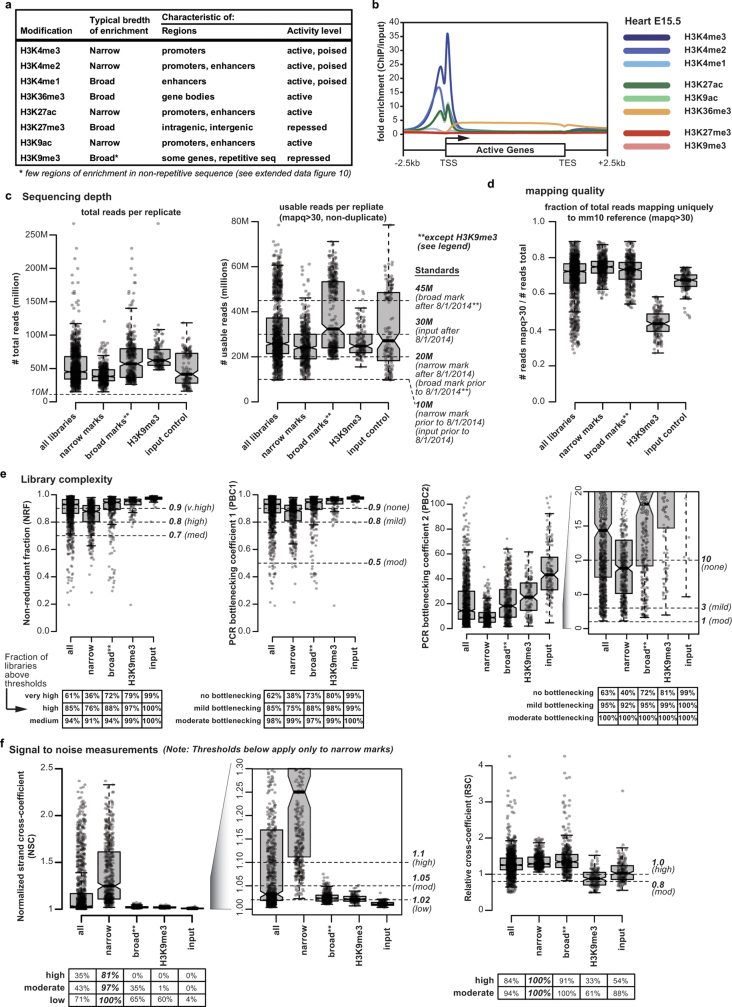
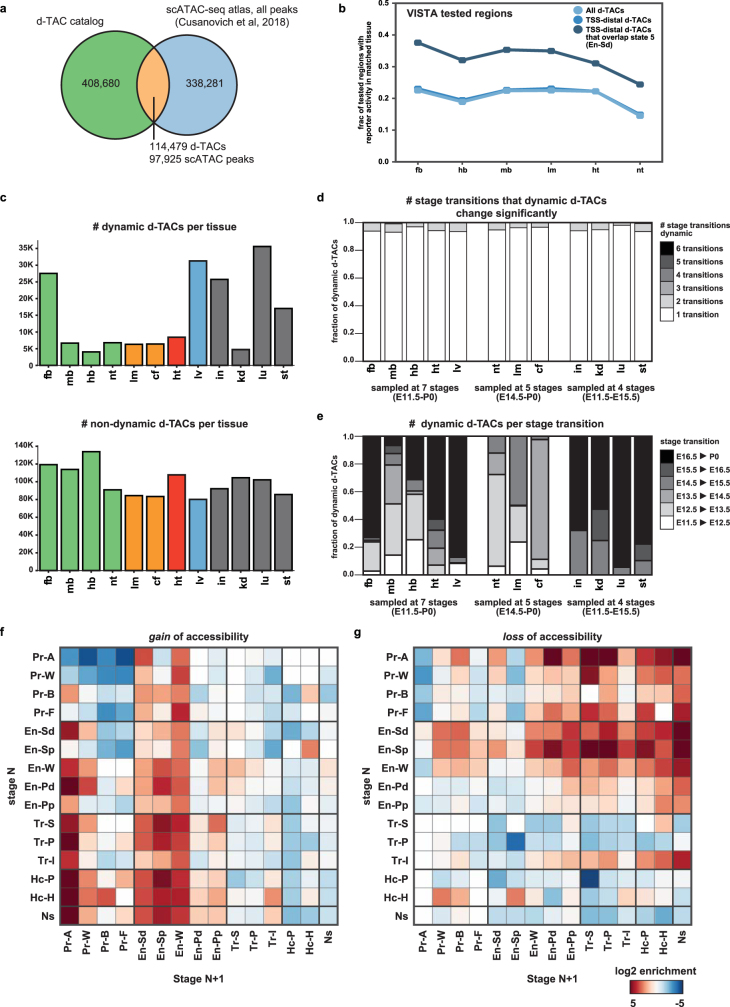
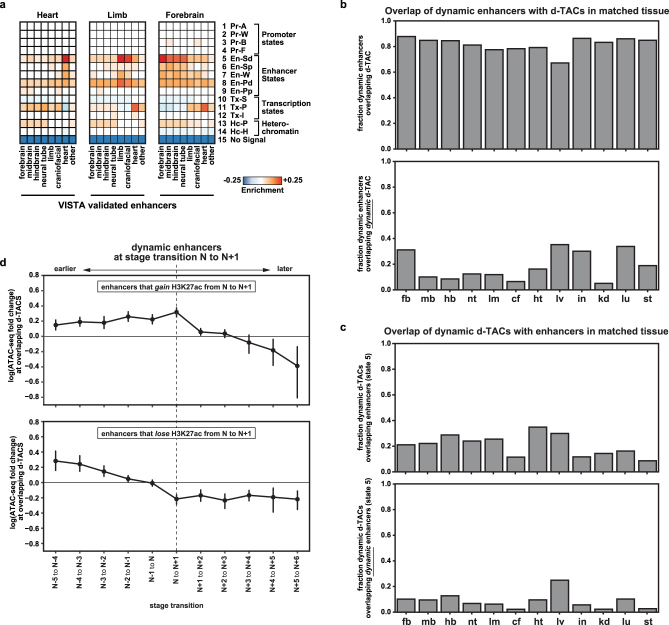
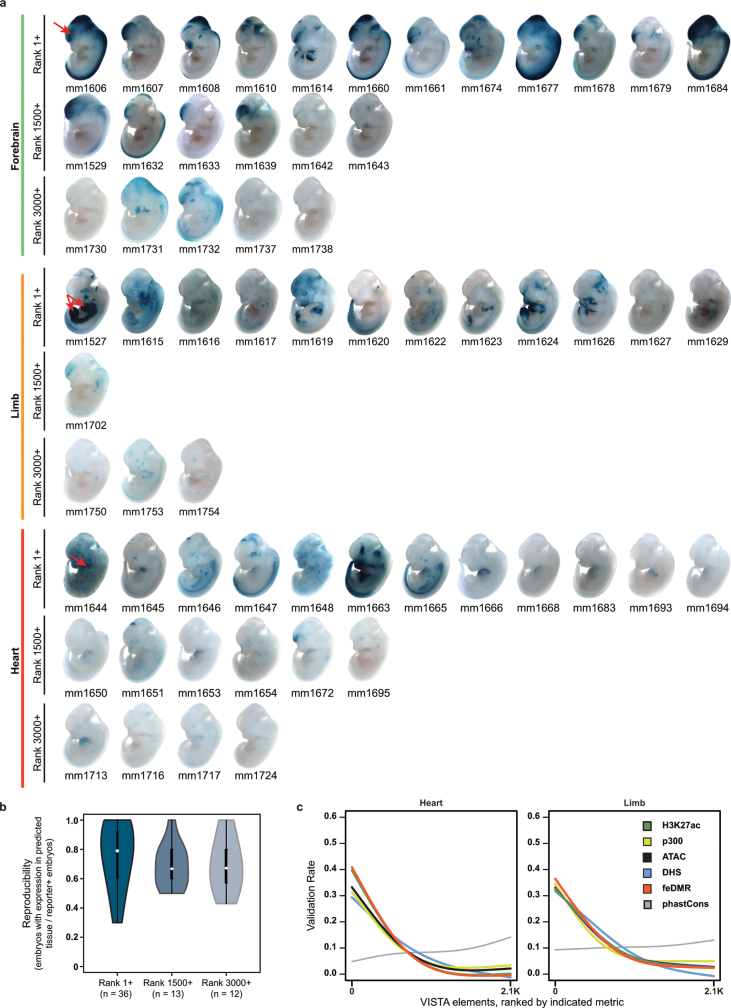
The Encyclopedia of DNA Elements (ENCODE) project has established a genomic resource for mammalian development, profiling a diverse panel of mouse tissues at 8 developmental stages from 10.5 days after conception until birth, including transcriptomes, methylomes and chromatin states. Here we systematically examined the state and accessibility of chromatin in the developing mouse fetus. In total we performed 1,128 chromatin immunoprecipitation with sequencing (ChIP-seq) assays for histone modifications and 132 assay for transposase-accessible chromatin using sequencing (ATAC-seq) assays for chromatin accessibility across 72 distinct tissue-stages. We used integrative analysis to develop a unified set of chromatin state annotations, infer the identities of dynamic enhancers and key transcriptional regulators, and characterize the relationship between chromatin state and accessibility during developmental gene regulation. We also leveraged these data to link enhancers to putative target genes and demonstrate tissue-specific enrichments of sequence variants associated with disease in humans. The mouse ENCODE data sets provide a compendium of resources for biomedical researchers and achieve, to our knowledge, the most comprehensive view of chromatin dynamics during mammalian fetal development to date.
Conflict of interest statement
B.R. is co-founder and share holder of Arima Genomics. The other authors declare no competing interests.
Figures

















References
-
- Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016). - PubMed
-
- Tessarz, P. & Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 (2014). - PubMed
-
- Shlyueva, D., Stampfel, G. & Stark, A. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
