

Spatiotemporal DNA methylome dynamics of the developing mouse fetus
- PMID: 32728242
- PMCID: PMC7398276
- DOI: 10.1038/s41586-020-2119-x
Spatiotemporal DNA methylome dynamics of the developing mouse fetus
Abstract
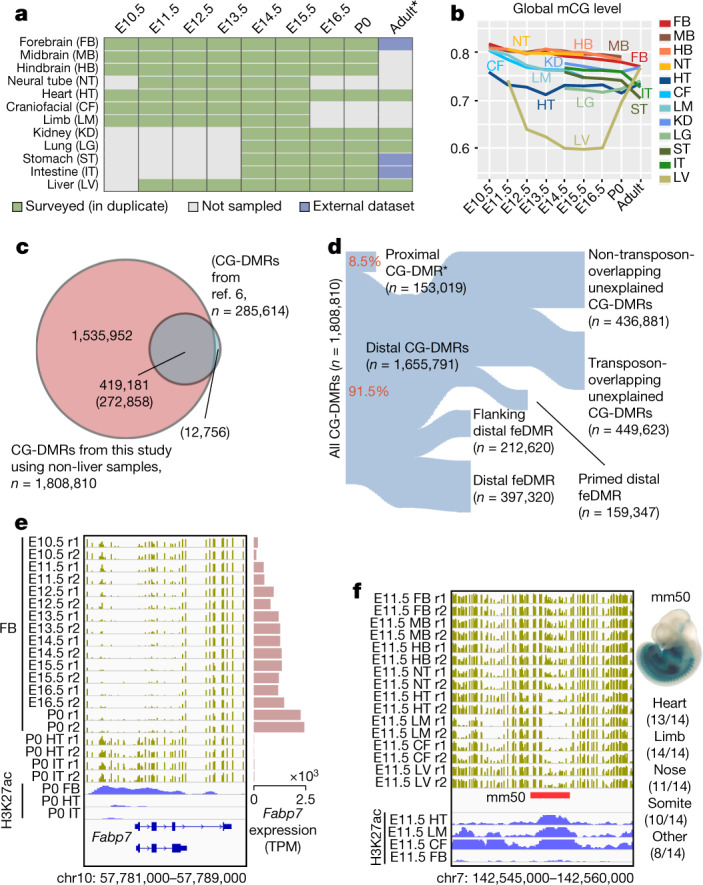
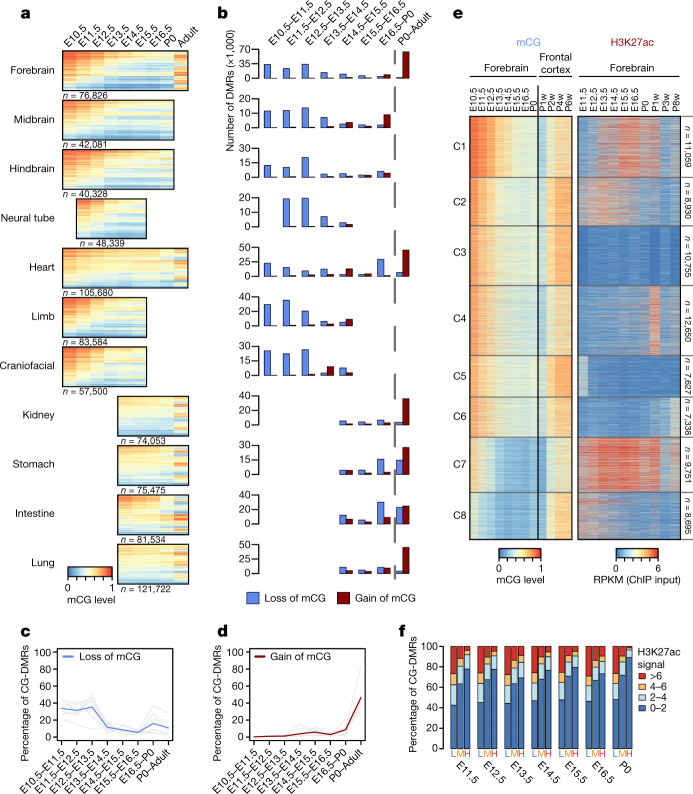
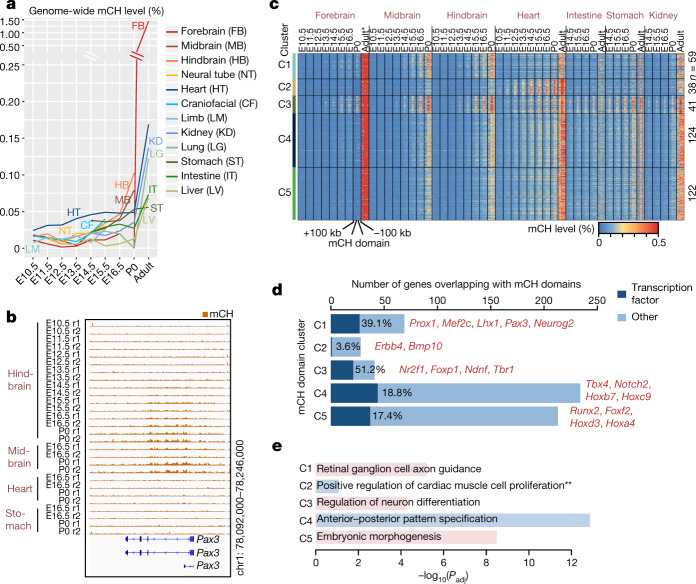
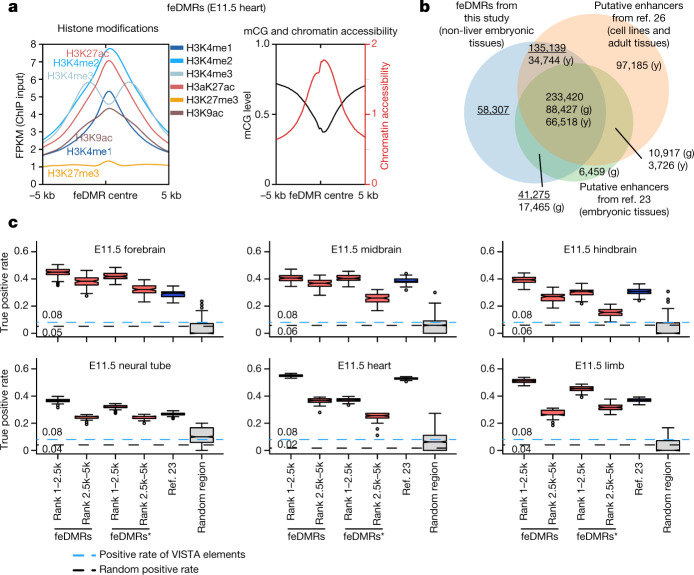
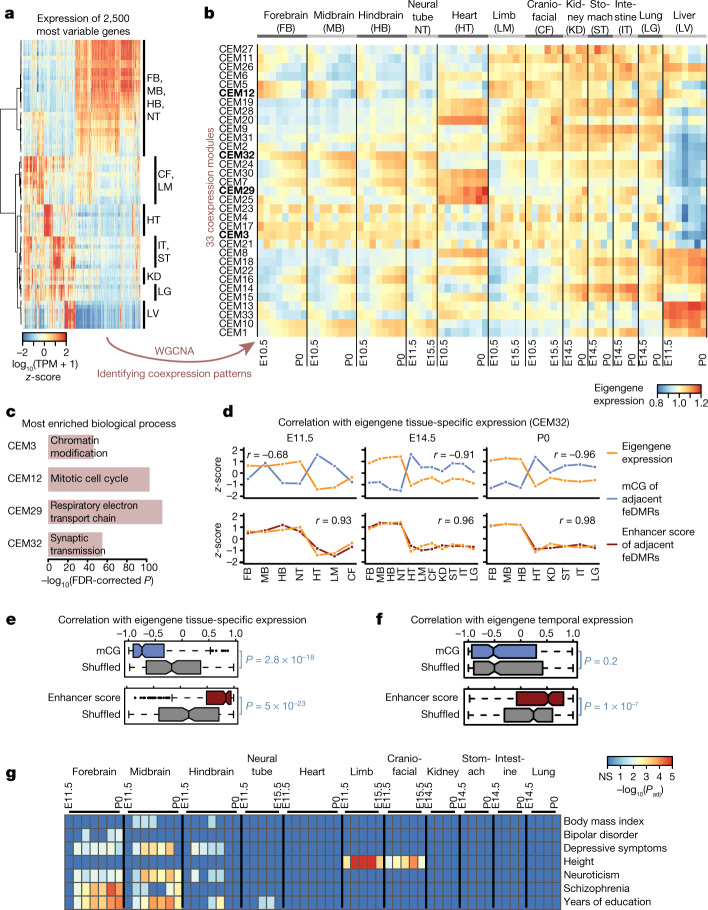
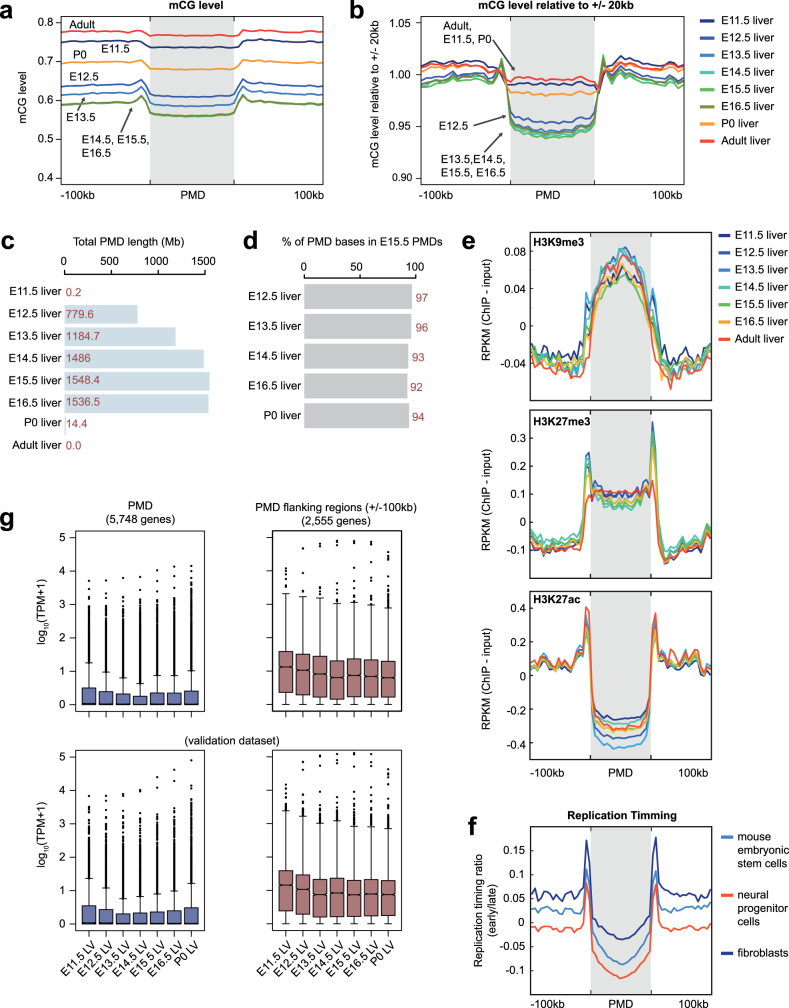
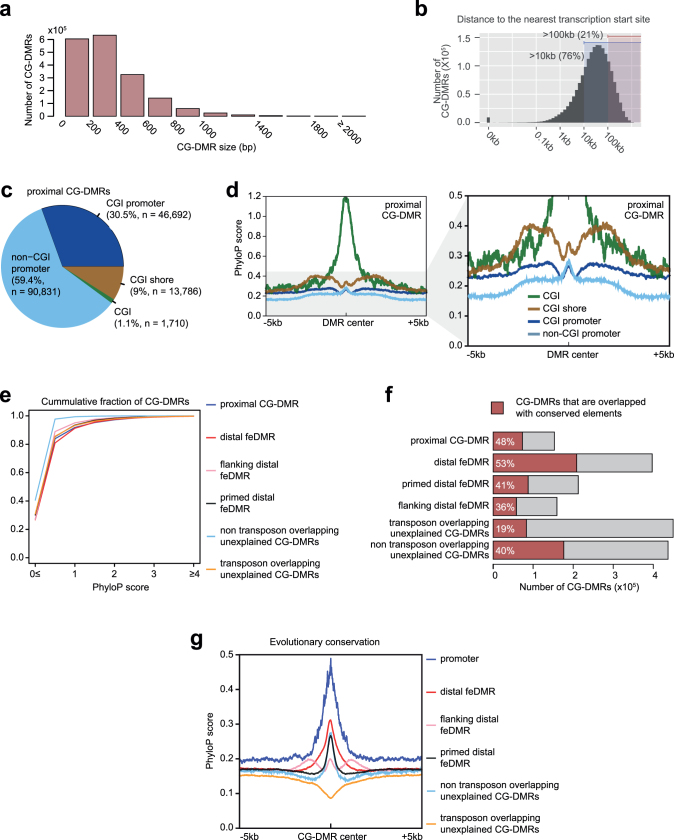
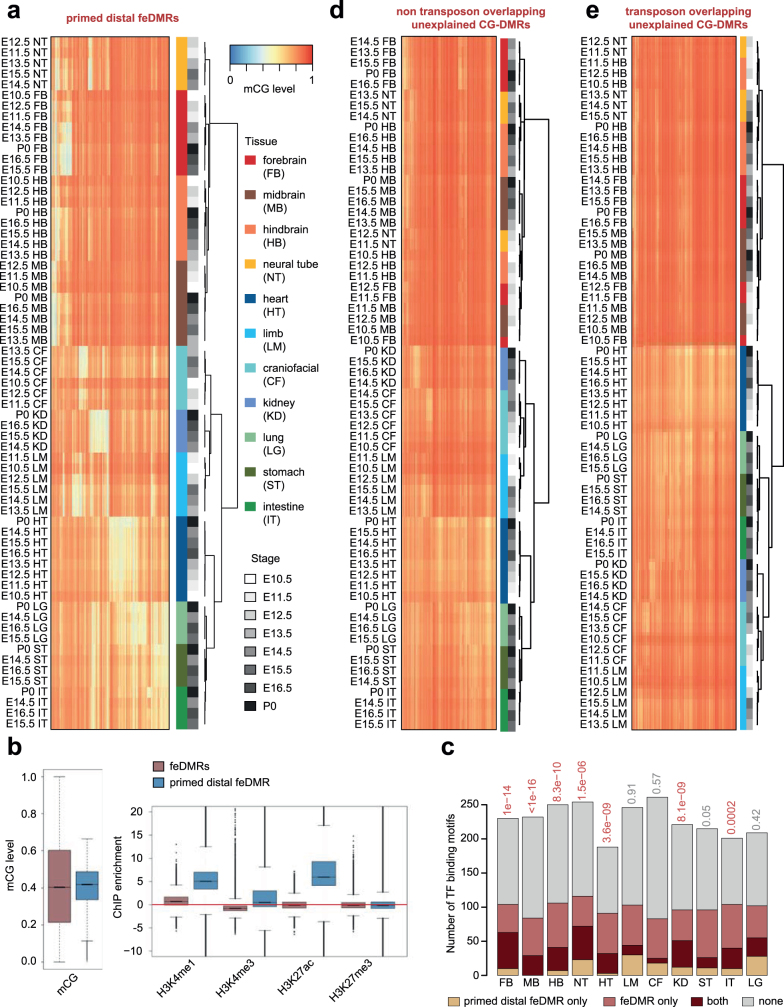
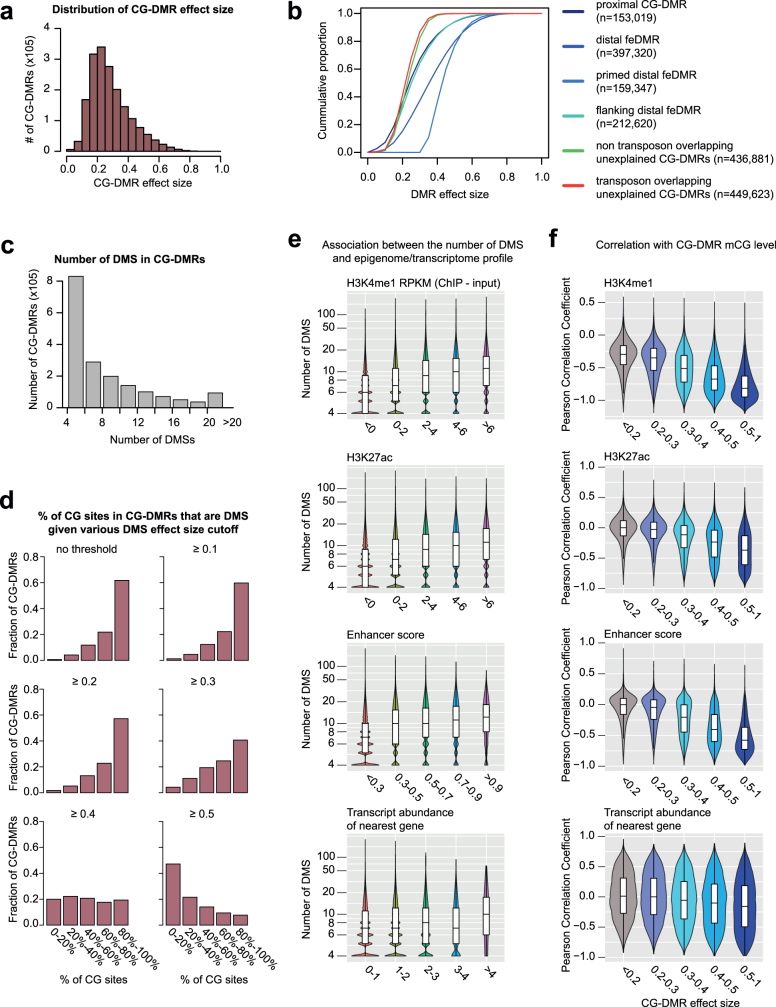
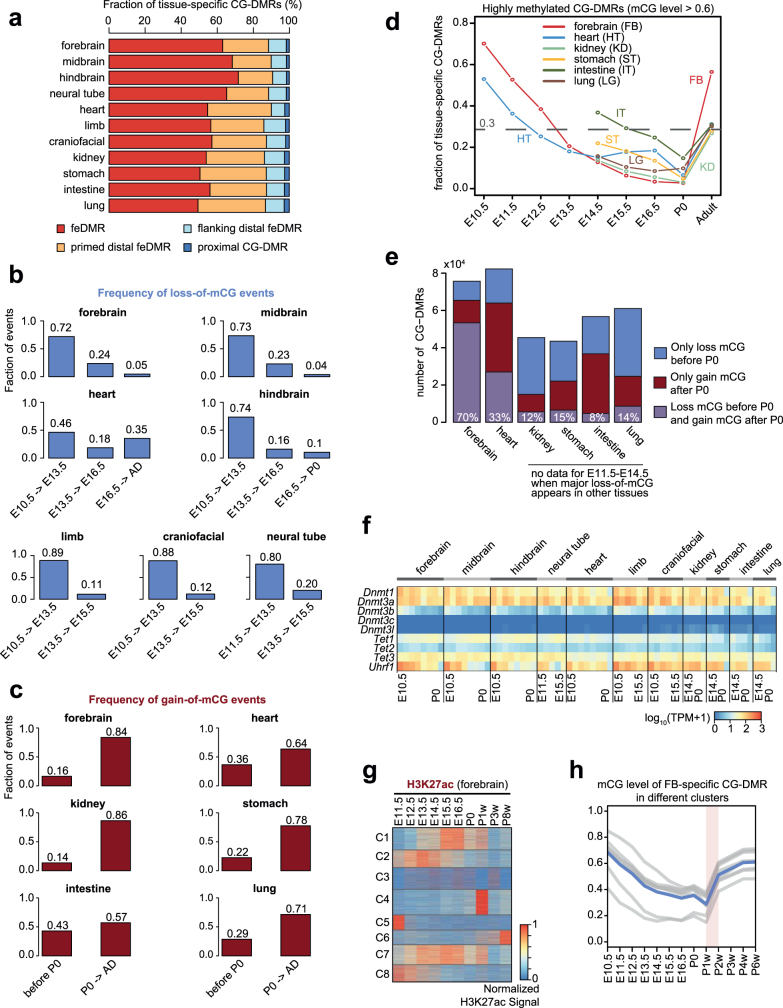
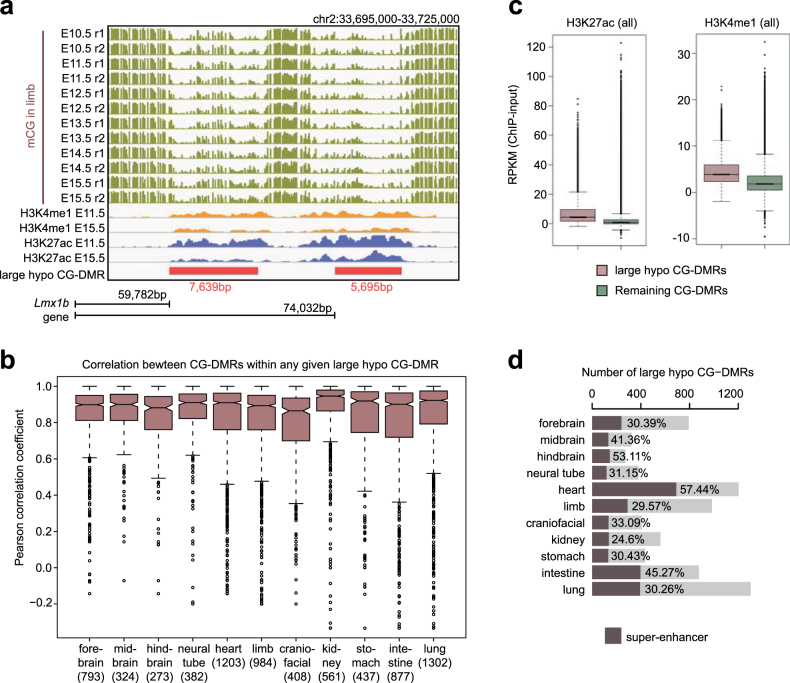
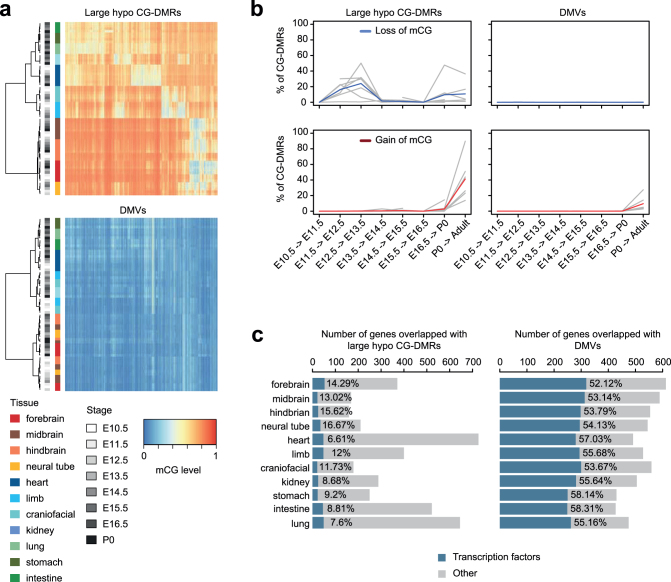
Cytosine DNA methylation is essential for mammalian development but understanding of its spatiotemporal distribution in the developing embryo remains limited1,2. Here, as part of the mouse Encyclopedia of DNA Elements (ENCODE) project, we profiled 168 methylomes from 12 mouse tissues or organs at 9 developmental stages from embryogenesis to adulthood. We identified 1,808,810 genomic regions that showed variations in CG methylation by comparing the methylomes of different tissues or organs from different developmental stages. These DNA elements predominantly lose CG methylation during fetal development, whereas the trend is reversed after birth. During late stages of fetal development, non-CG methylation accumulated within the bodies of key developmental transcription factor genes, coinciding with their transcriptional repression. Integration of genome-wide DNA methylation, histone modification and chromatin accessibility data enabled us to predict 461,141 putative developmental tissue-specific enhancers, the human orthologues of which were enriched for disease-associated genetic variants. These spatiotemporal epigenome maps provide a resource for studies of gene regulation during tissue or organ progression, and a starting point for investigating regulatory elements that are involved in human developmental disorders.
Conflict of interest statement
B.R. is a co-founder and shareholder of Arima Genomics, Inc. The other authors declare no competing interests.
Figures
References
-
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature447, 425–432 (2007). - PubMed
-
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002). - PubMed
-
- Spitz, F. & Furlong, E. E. M. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
