

Occupancy maps of 208 chromatin-associated proteins in one human cell type
- PMID: 32728244
- PMCID: PMC7398277
- DOI: 10.1038/s41586-020-2023-4
Occupancy maps of 208 chromatin-associated proteins in one human cell type
Abstract
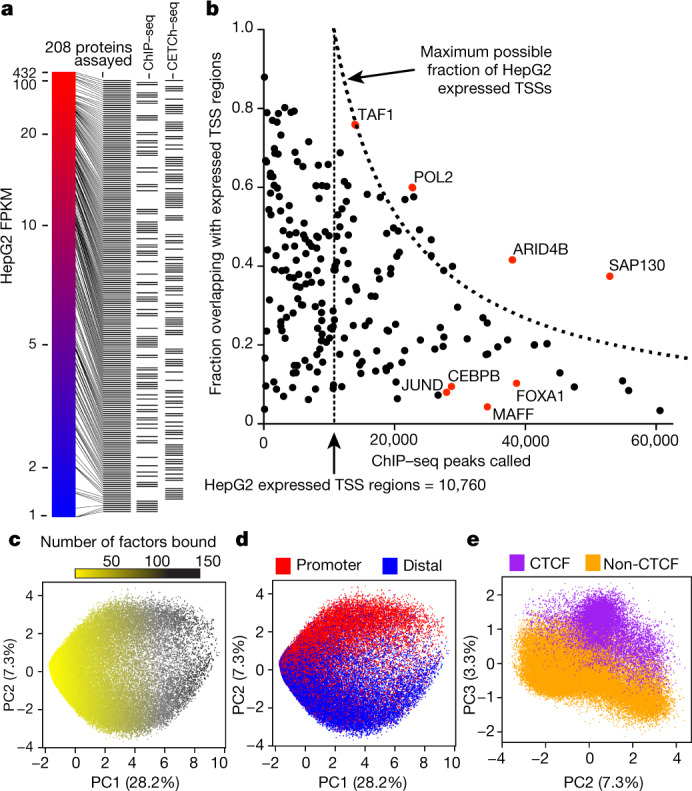
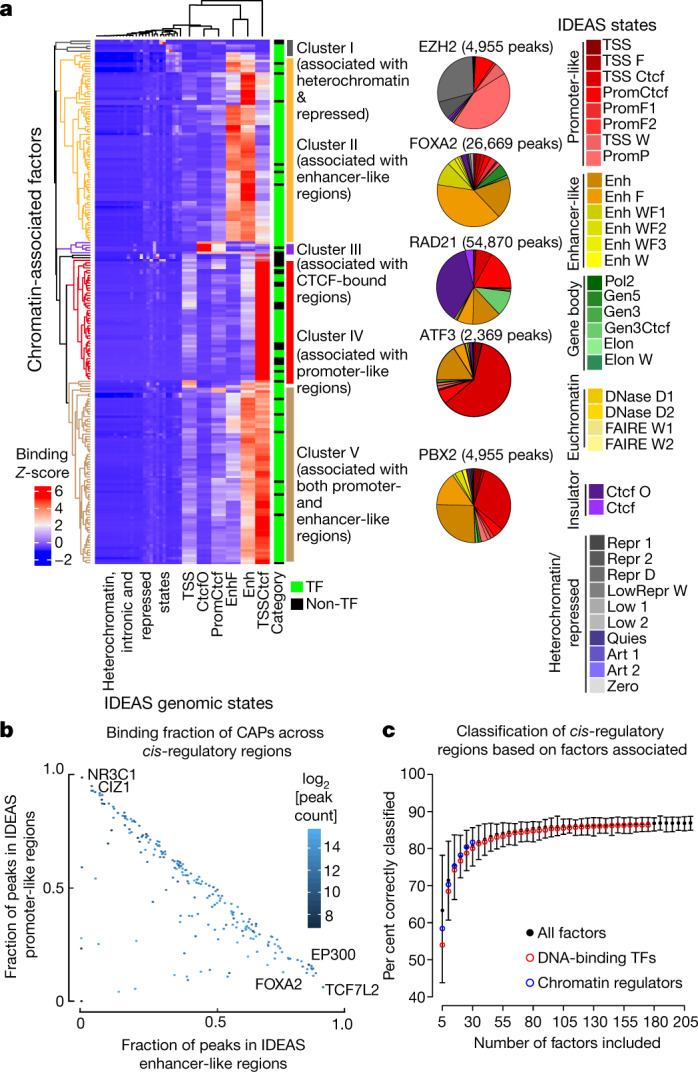
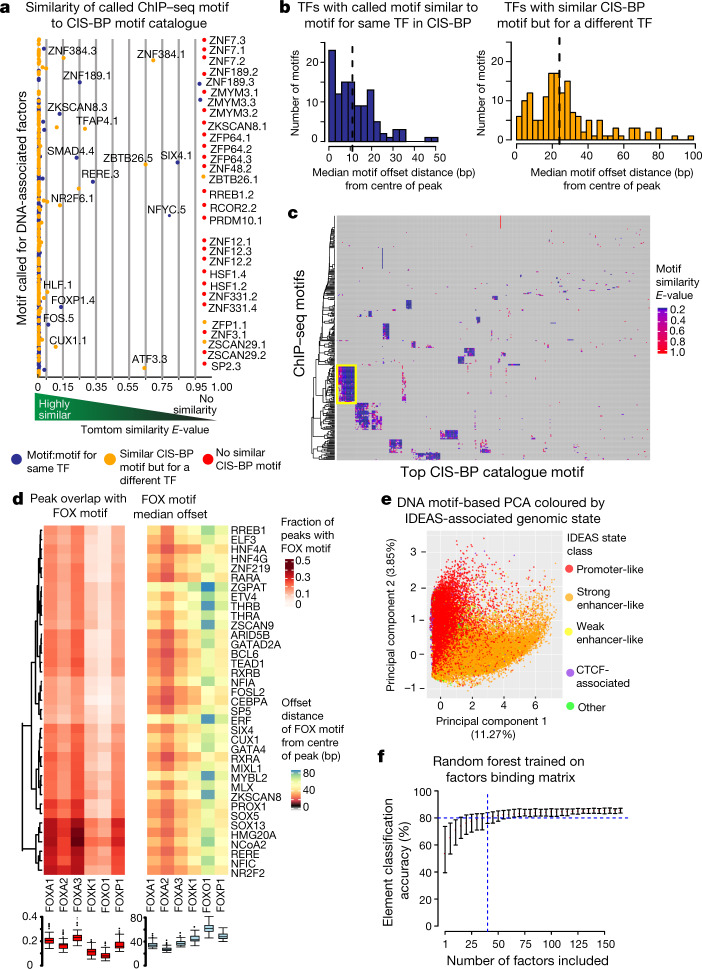
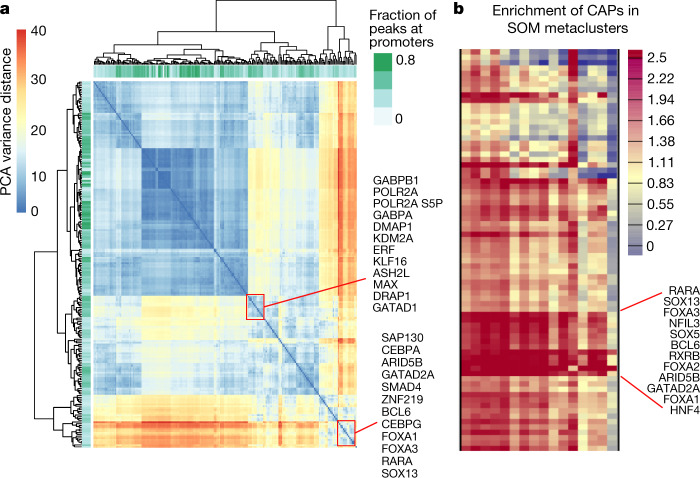
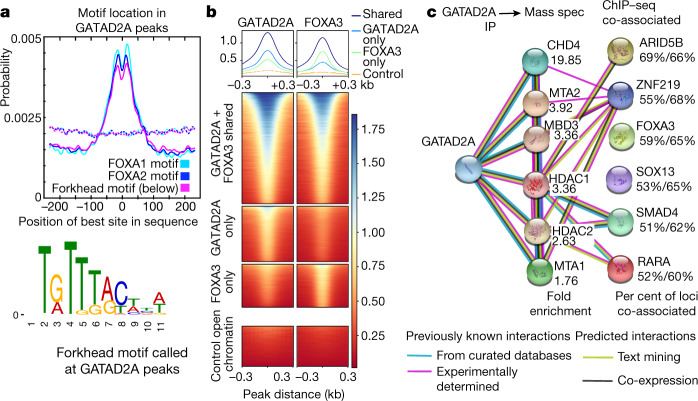
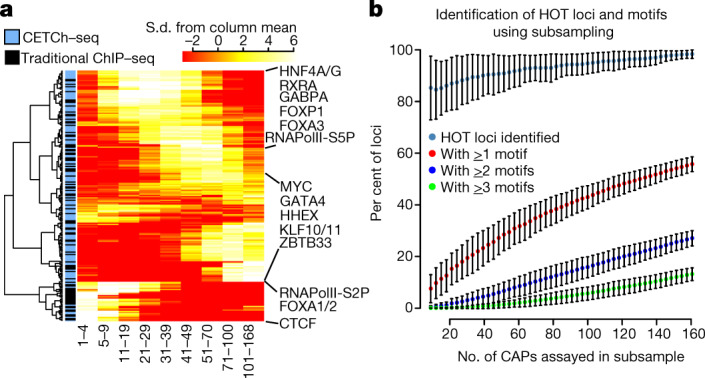
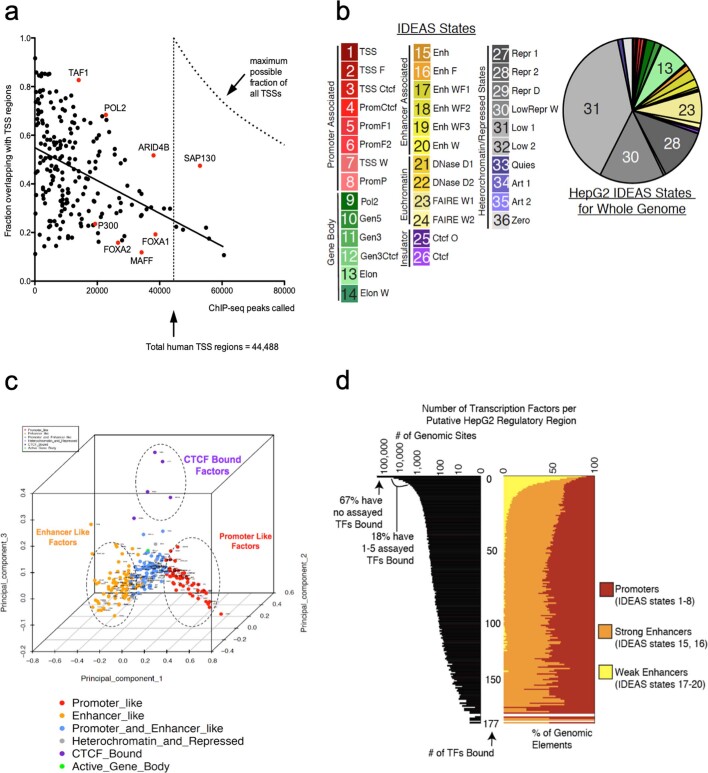
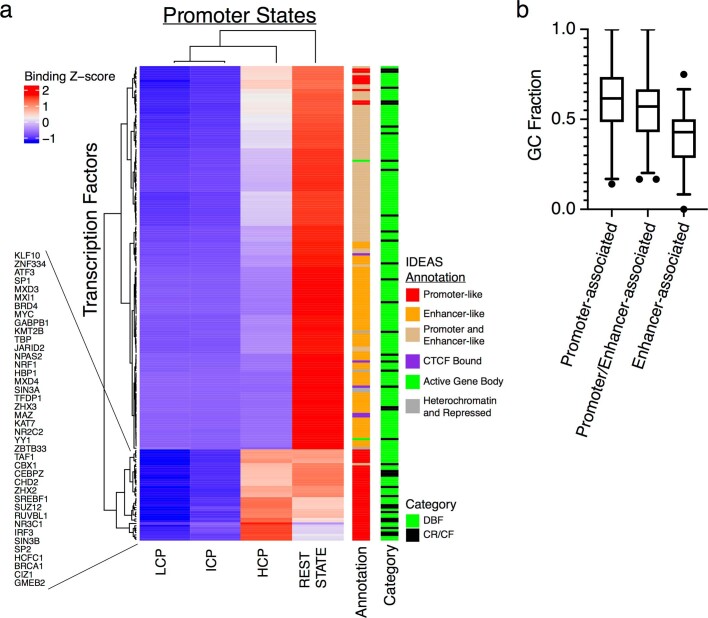
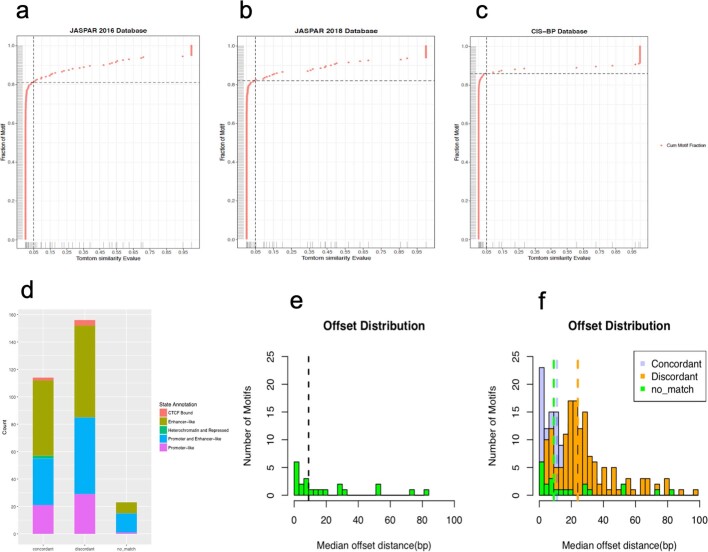
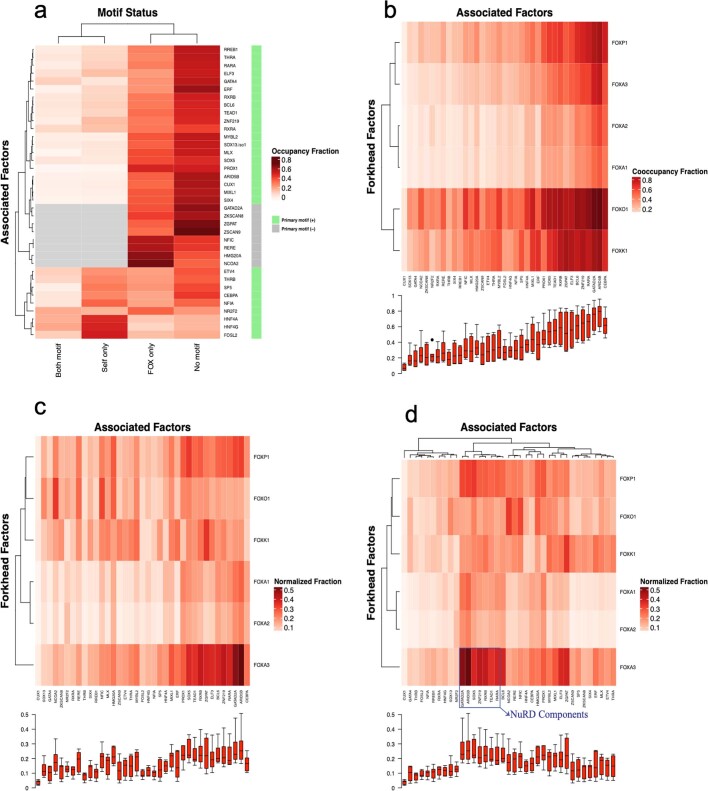
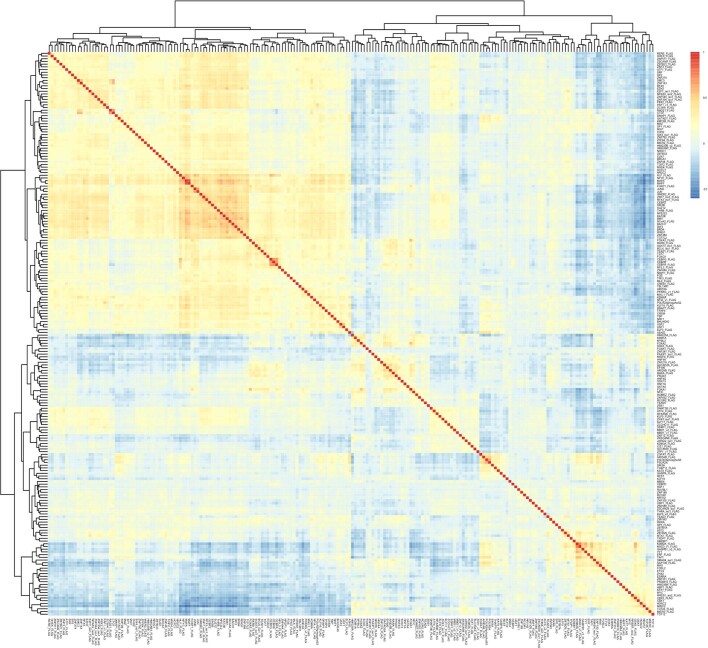
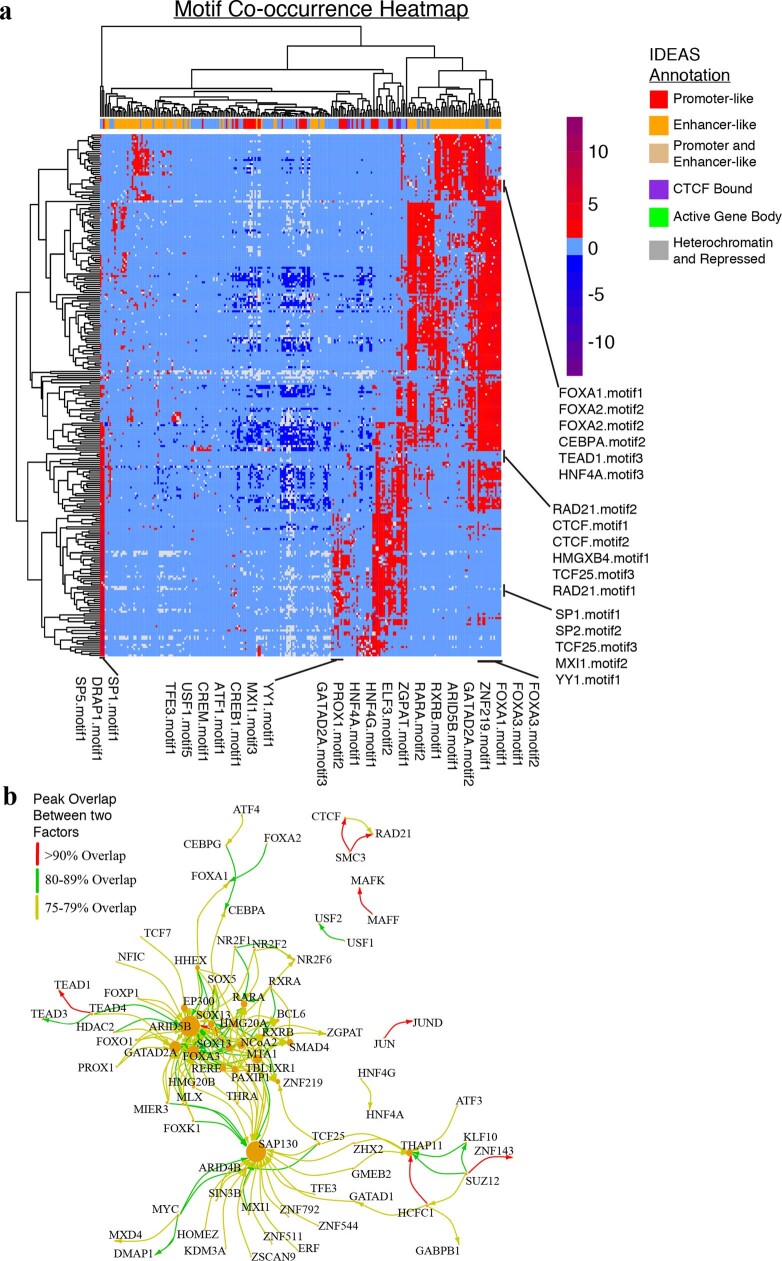
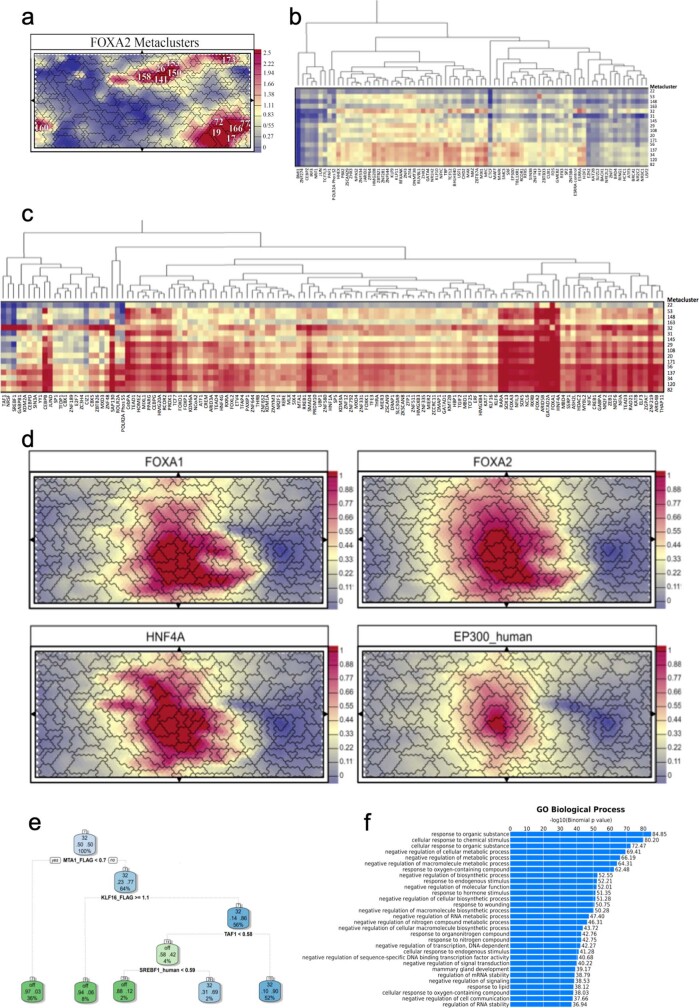
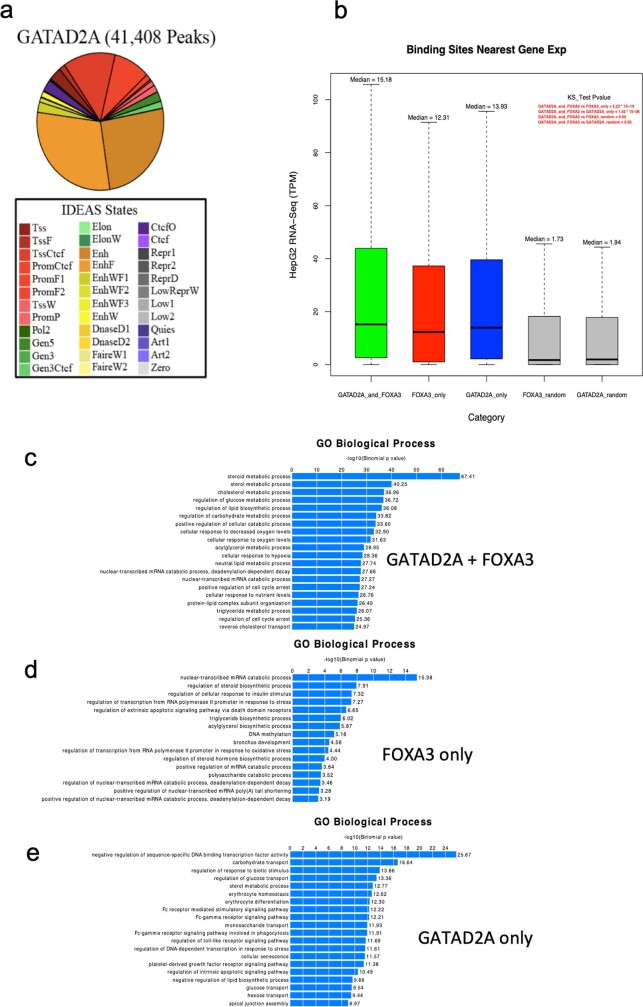
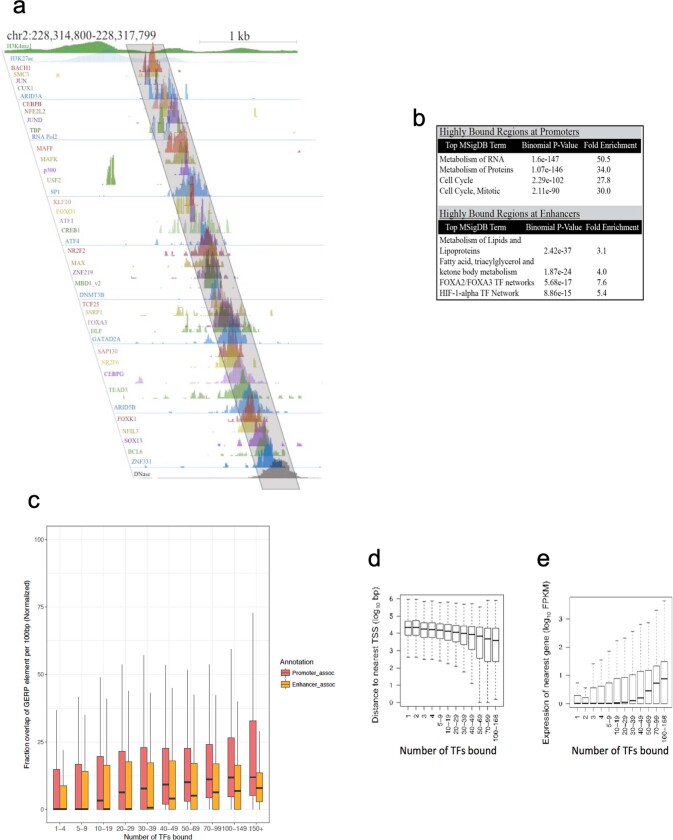
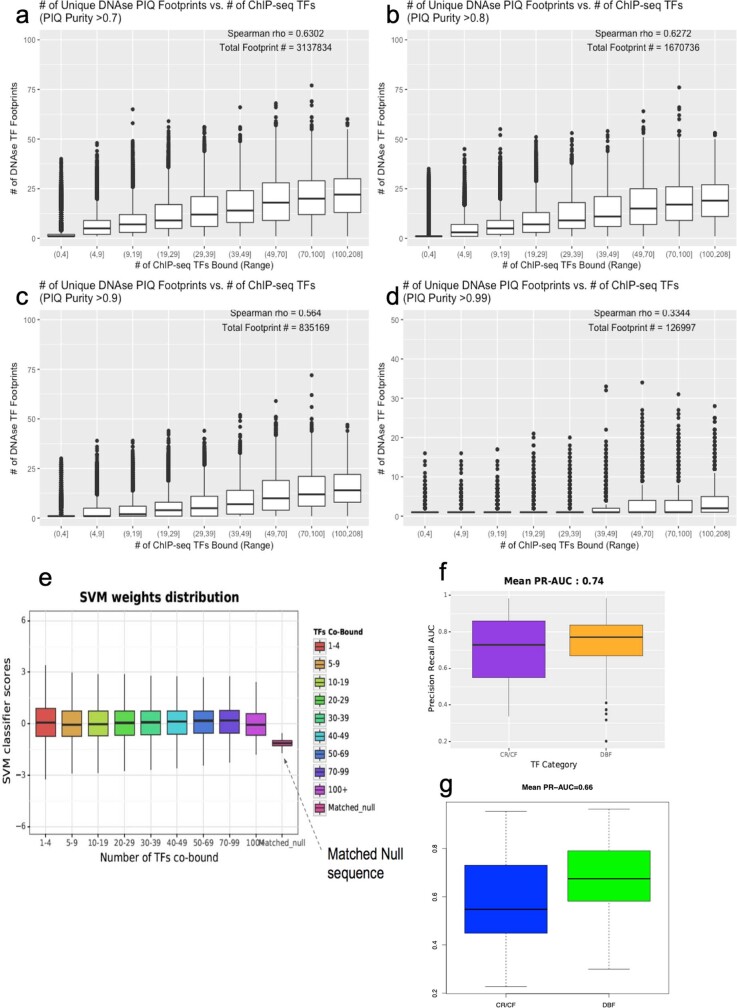
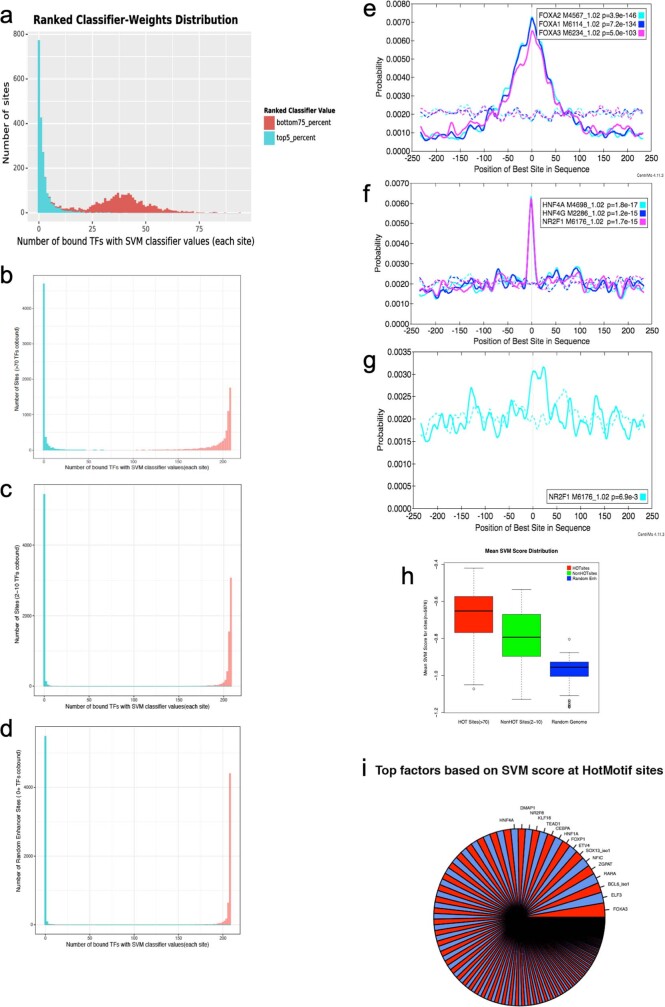
Transcription factors are DNA-binding proteins that have key roles in gene regulation1,2. Genome-wide occupancy maps of transcriptional regulators are important for understanding gene regulation and its effects on diverse biological processes3-6. However, only a minority of the more than 1,600 transcription factors encoded in the human genome has been assayed. Here we present, as part of the ENCODE (Encyclopedia of DNA Elements) project, data and analyses from chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) experiments using the human HepG2 cell line for 208 chromatin-associated proteins (CAPs). These comprise 171 transcription factors and 37 transcriptional cofactors and chromatin regulator proteins, and represent nearly one-quarter of CAPs expressed in HepG2 cells. The binding profiles of these CAPs form major groups associated predominantly with promoters or enhancers, or with both. We confirm and expand the current catalogue of DNA sequence motifs for transcription factors, and describe motifs that correspond to other transcription factors that are co-enriched with the primary ChIP target. For example, FOX family motifs are enriched in ChIP-seq peaks of 37 other CAPs. We show that motif content and occupancy patterns can distinguish between promoters and enhancers. This catalogue reveals high-occupancy target regions at which many CAPs associate, although each contains motifs for only a minority of the numerous associated transcription factors. These analyses provide a more complete overview of the gene regulatory networks that define this cell type, and demonstrate the usefulness of the large-scale production efforts of the ENCODE Consortium.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Vaquerizas, J. M., Kummerfeld, S. K., Teichmann, S. A. & Luscombe, N. M. A census of human transcription factors: function, expression and evolution. Nat. Rev. Genet. 10, 252–263 (2009). - PubMed
-
- Lambert, S. A. et al. The human transcription factors. Cell172, 650–665 (2018). - PubMed
-
- Chen, X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell133, 1106–1117 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
