

An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells
- PMID: 32730809
- PMCID: PMC7775622
- DOI: 10.1016/j.cell.2020.07.001
An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells
Abstract
Electrophilic compounds originating from nature or chemical synthesis have profound effects on immune cells. These compounds are thought to act by cysteine modification to alter the functions of immune-relevant proteins; however, our understanding of electrophile-sensitive cysteines in the human immune proteome remains limited. Here, we present a global map of cysteines in primary human T cells that are susceptible to covalent modification by electrophilic small molecules. More than 3,000 covalently liganded cysteines were found on functionally and structurally diverse proteins, including many that play fundamental roles in immunology. We further show that electrophilic compounds can impair T cell activation by distinct mechanisms involving the direct functional perturbation and/or degradation of proteins. Our findings reveal a rich content of ligandable cysteines in human T cells and point to electrophilic small molecules as a fertile source for chemical probes and ultimately therapeutics that modulate immunological processes and their associated disorders.
Keywords: BIRC3; ITK; T cells; activity-based protein profiling; chemical proteomics; covalent; cysteine; electrophiles; human; protein degradation.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests B.F.C. is a founder and scientific advisor to Vividion Therapeutics. B.F.C., V.M.C., B.M., D.R., M.A.S., E.V.V., X.Z., and M.Y. are co-inventors on a patent application related to this work.
Figures
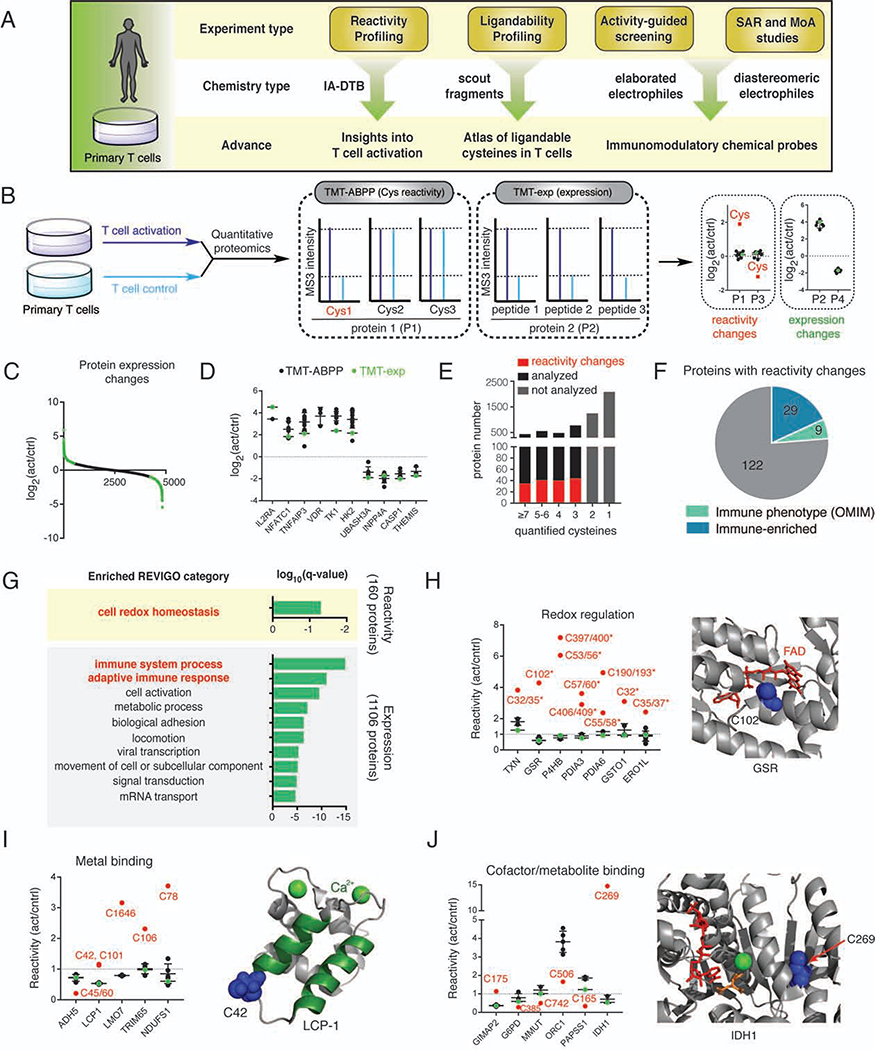
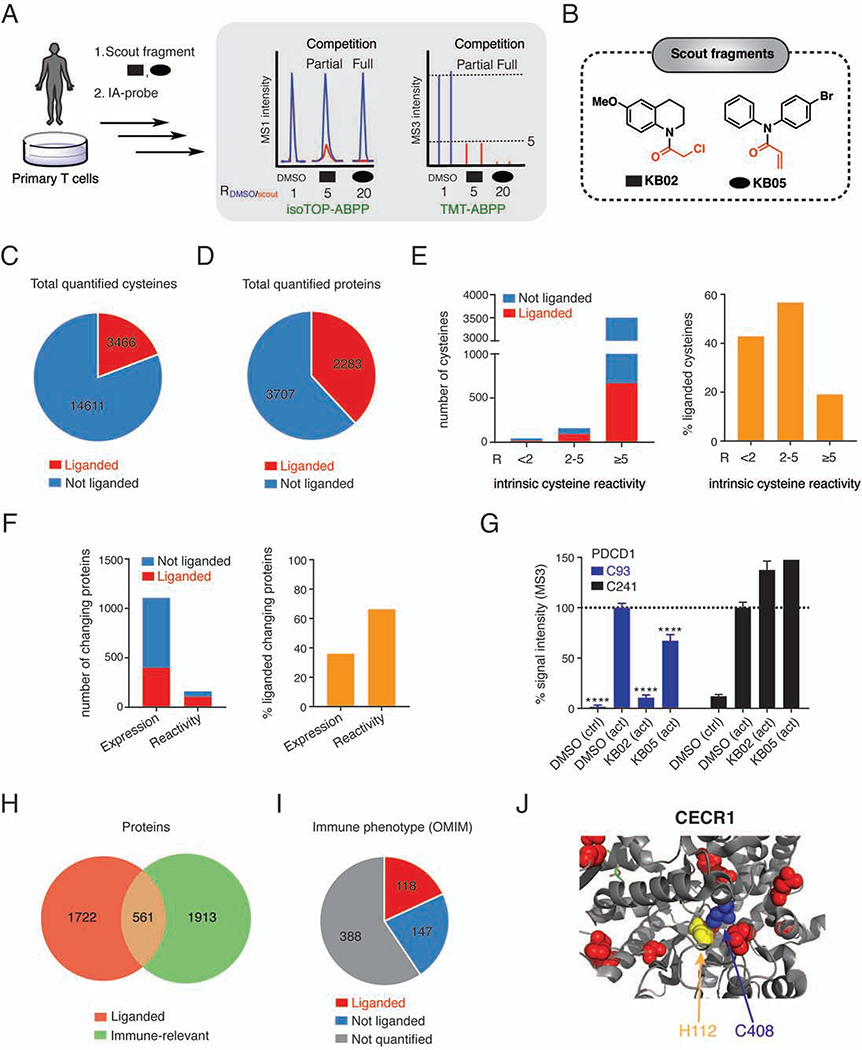
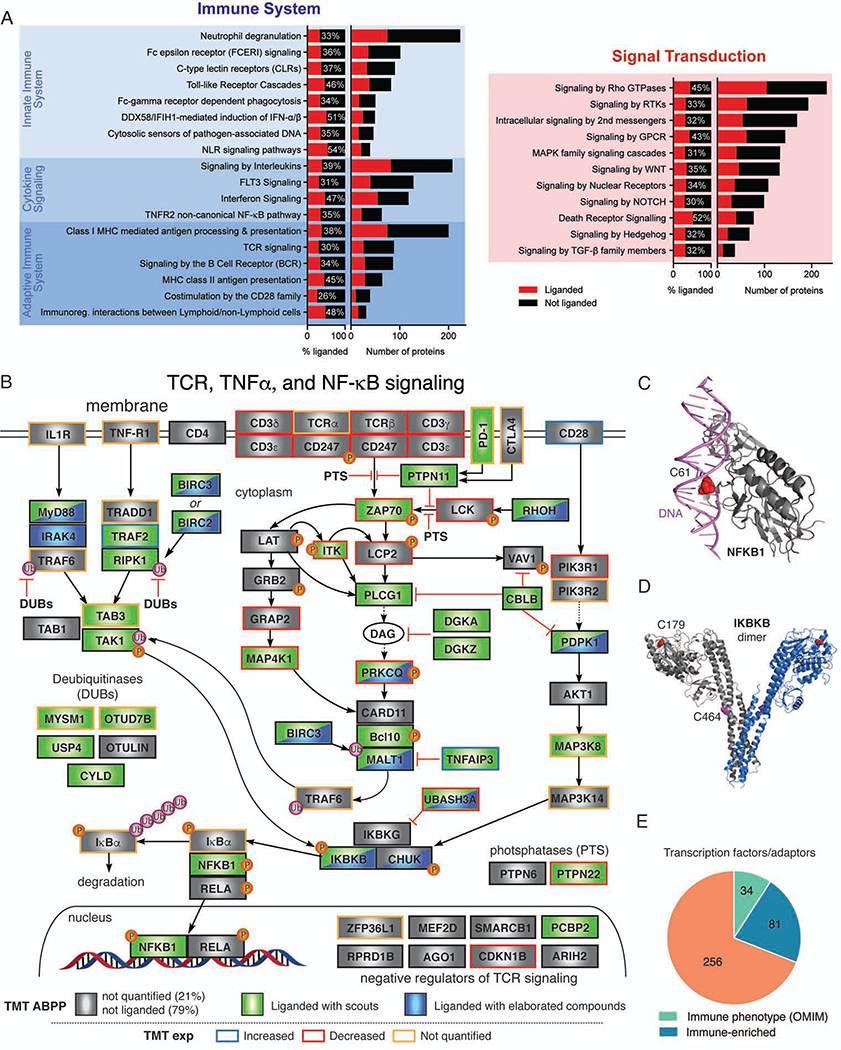
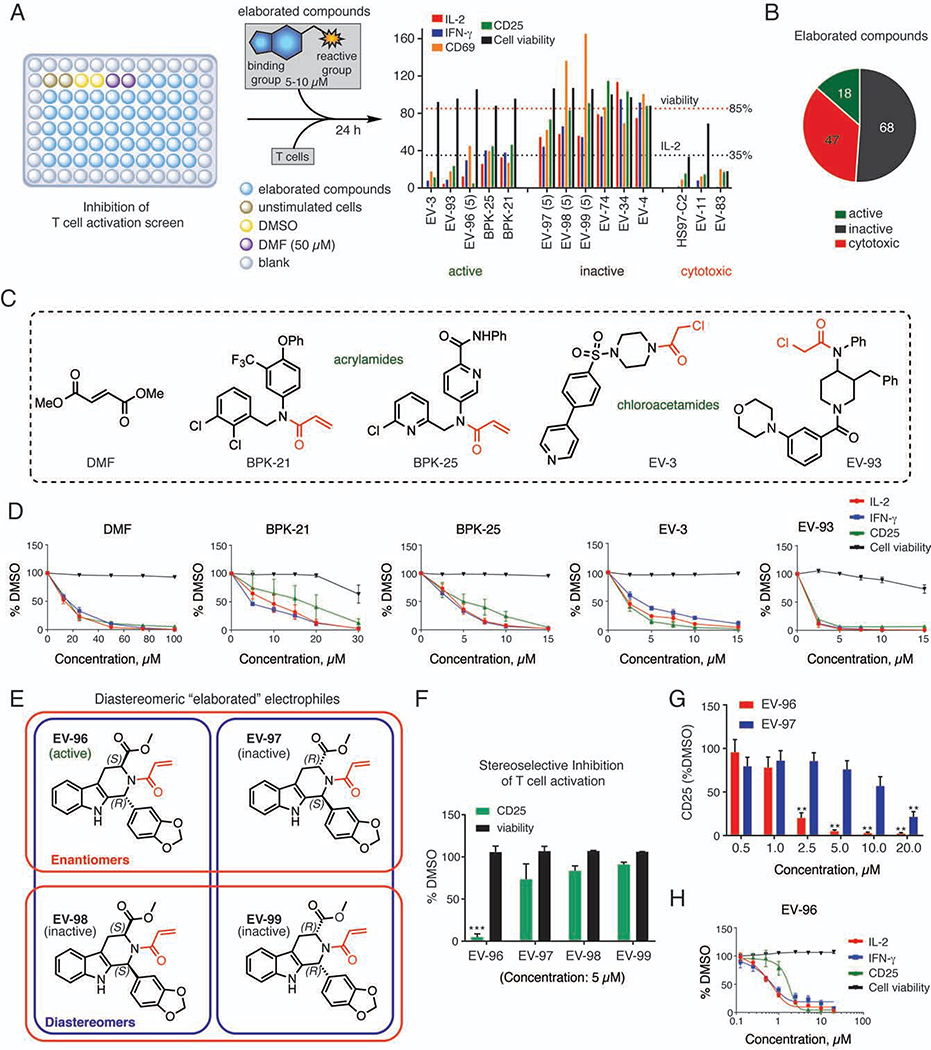
References
-
- Acuto O, and Cantrell D (2000). T cell activation and the cytoskeleton. Annu Rev Immunol 18, 165–184. - PubMed
-
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, and Honjo T (1996). Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. International immunology 8, 765–772. - PubMed
-
- Almeida L, Lochner M, Berod L, and Sparwasser T (2016). Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol 28, 514–524. - PubMed
-
- Andreotti AH, Joseph RE, Conley JM, Iwasa J, and Berg LJ (2018). Multidomain Control Over TEC Kinase Activation State Tunes the T Cell Response. Annu Rev Immunol 36, 549–578. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
