

Inhibition of miR-1193 leads to synthetic lethality in glioblastoma multiforme cells deficient of DNA-PKcs
- PMID: 32732911
- PMCID: PMC7393494
- DOI: 10.1038/s41419-020-02812-3
Inhibition of miR-1193 leads to synthetic lethality in glioblastoma multiforme cells deficient of DNA-PKcs
Abstract
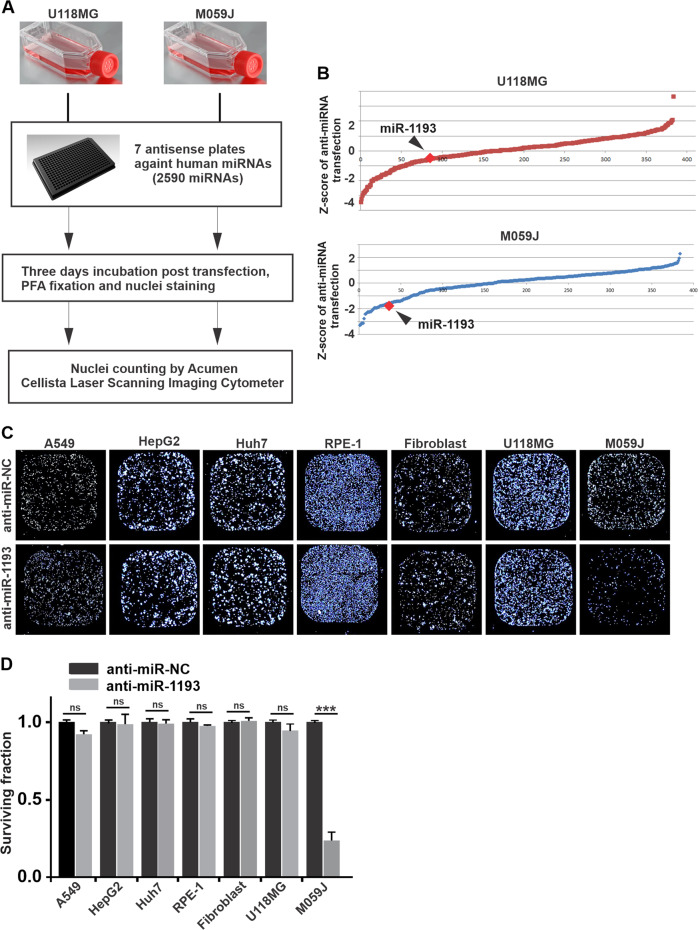
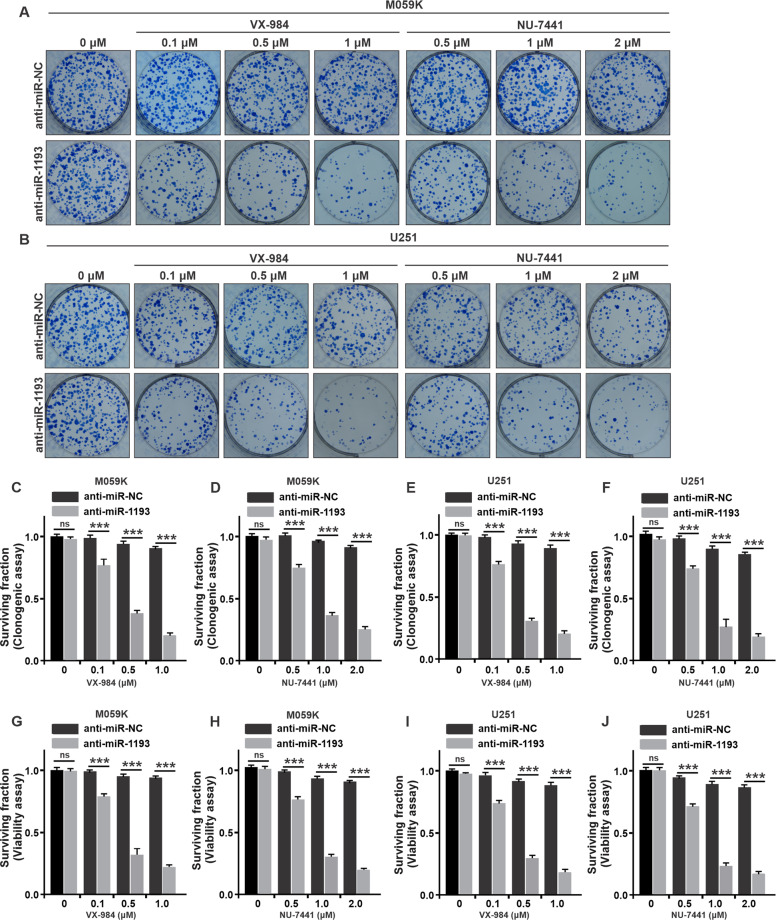
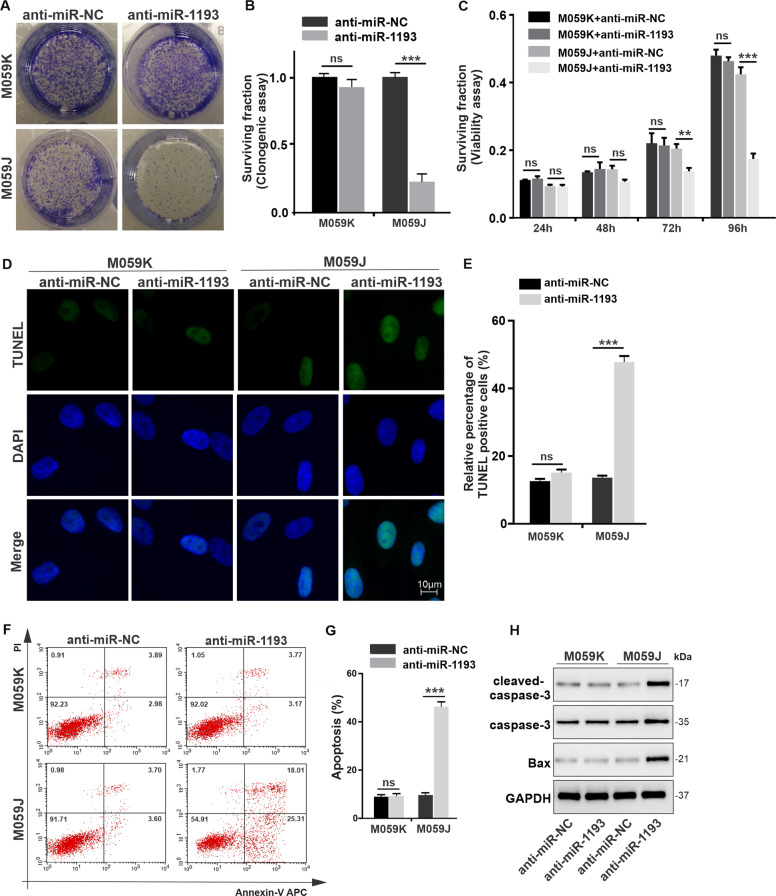
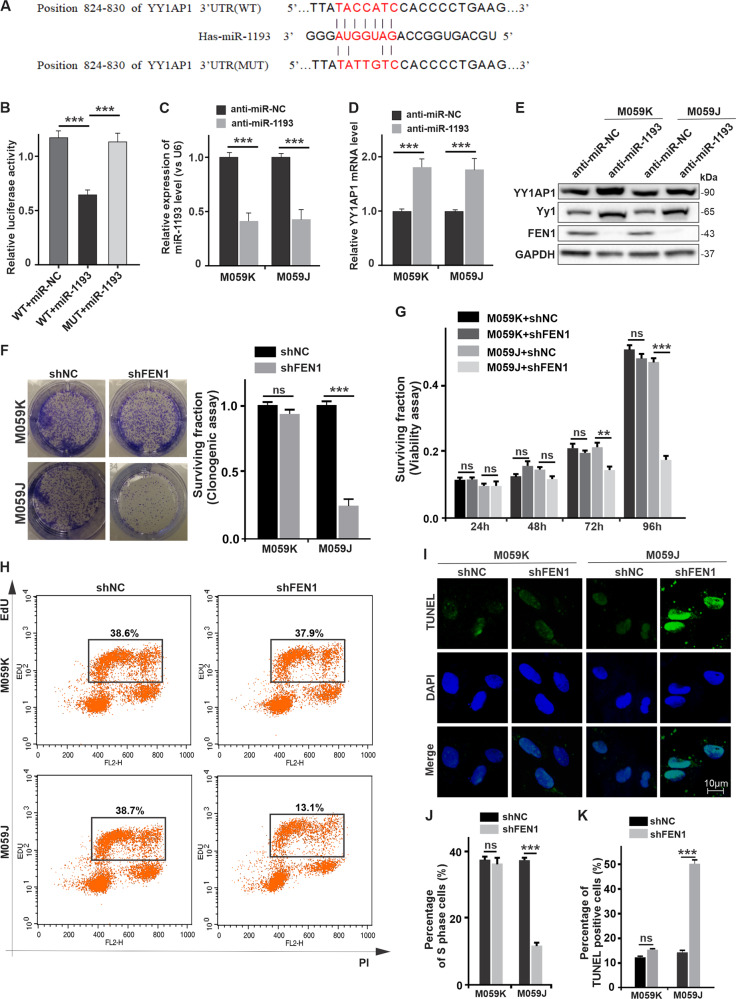
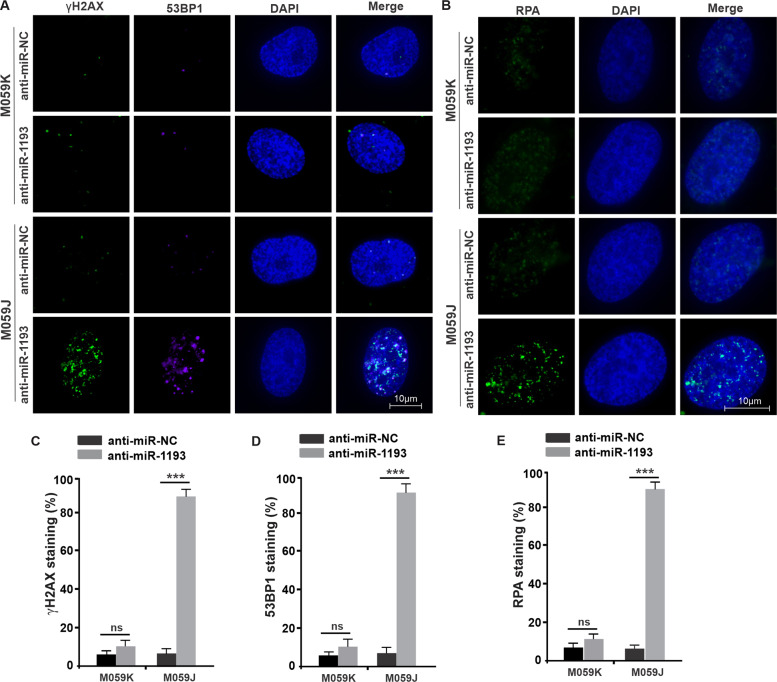
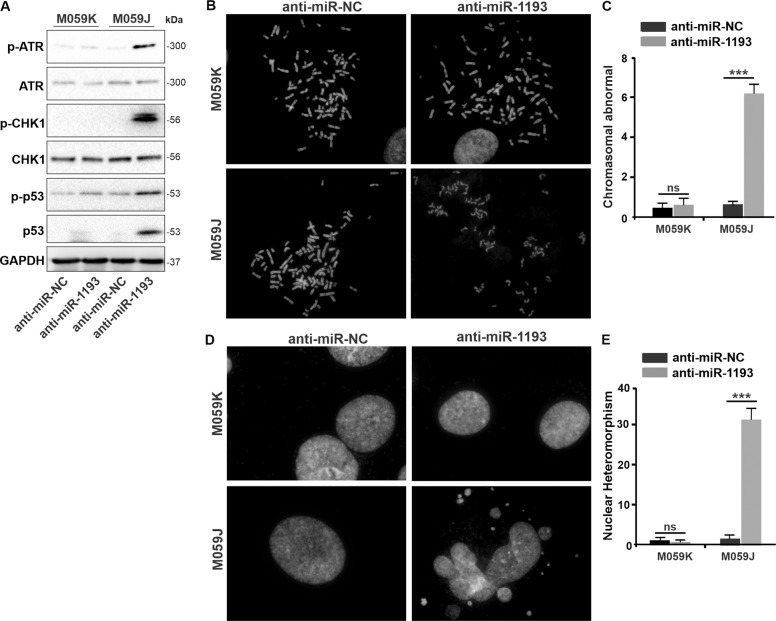
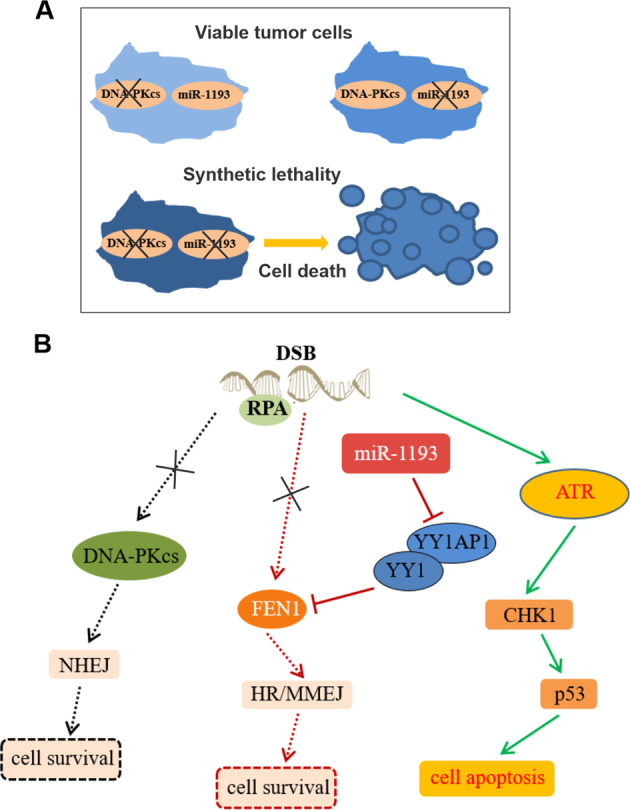
Glioblastoma multiforme (GBM) is the most malignant primary brain tumor and has the highest mortality rate among cancers and high resistance to radiation and cytotoxic chemotherapy. Although some targeted therapies can partially inhibit oncogenic mutation-driven proliferation of GBM cells, therapies harnessing synthetic lethality are 'coincidental' treatments with high effectiveness in cancers with gene mutations, such as GBM, which frequently exhibits DNA-PKcs mutation. By implementing a highly efficient high-throughput screening (HTS) platform using an in-house-constructed genome-wide human microRNA inhibitor library, we demonstrated that miR-1193 inhibition sensitized GBM tumor cells with DNA-PKcs deficiency. Furthermore, we found that miR-1193 directly targets YY1AP1, leading to subsequent inhibition of FEN1, an important factor in DNA damage repair. Inhibition of miR-1193 resulted in accumulation of DNA double-strand breaks and thus increased genomic instability. RPA-coated ssDNA structures enhanced ATR checkpoint kinase activity, subsequently activating the CHK1/p53/apoptosis axis. These data provide a preclinical theory for the application of miR-1193 inhibition as a potential synthetic lethal approach targeting GBM cancer cells with DNA-PKcs deficiency.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition.Oncotarget. 2015 Oct 20;6(32):32396-409. doi: 10.18632/oncotarget.6136. Oncotarget. 2015. PMID: 26486089 Free PMC article.
-
SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors.Proc Natl Acad Sci U S A. 2021 Feb 9;118(6):e2015654118. doi: 10.1073/pnas.2015654118. Proc Natl Acad Sci U S A. 2021. PMID: 33536335 Free PMC article.
-
Depletion of ATR selectively sensitizes ATM-deficient human mammary epithelial cells to ionizing radiation and DNA-damaging agents.Cell Cycle. 2014;13(22):3541-50. doi: 10.4161/15384101.2014.960729. Cell Cycle. 2014. PMID: 25483091 Free PMC article.
-
Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma.Int J Mol Sci. 2020 Jul 11;21(14):4910. doi: 10.3390/ijms21144910. Int J Mol Sci. 2020. PMID: 32664581 Free PMC article. Review.
-
microRNAs: Potential glioblastoma radiosensitizer by targeting radiation-related molecular pathways.Mutat Res. 2019 Nov;816-818:111679. doi: 10.1016/j.mrfmmm.2019.111679. Epub 2019 Oct 21. Mutat Res. 2019. PMID: 31715522 Review.
Cited by
-
Potential promising of synthetic lethality in cancer research and treatment.Naunyn Schmiedebergs Arch Pharmacol. 2025 Feb;398(2):1403-1431. doi: 10.1007/s00210-024-03444-6. Epub 2024 Sep 21. Naunyn Schmiedebergs Arch Pharmacol. 2025. PMID: 39305329 Review.
-
Small-Molecule Inhibitors Targeting FEN1 for Cancer Therapy.Biomolecules. 2022 Jul 20;12(7):1007. doi: 10.3390/biom12071007. Biomolecules. 2022. PMID: 35883563 Free PMC article. Review.
-
Integrated analysis of differential expression profile of miRNA in the uterus of seasonal estrus sheep.BMC Genomics. 2025 Mar 13;26(1):243. doi: 10.1186/s12864-025-11401-7. BMC Genomics. 2025. PMID: 40082757 Free PMC article.
-
microRNA-497-5p-based screening identifies a novel synthetic lethal-type interaction via PKMYT1 and WEE1 in pleural mesothelioma.Mol Ther Nucleic Acids. 2025 Jun 17;36(3):102610. doi: 10.1016/j.omtn.2025.102610. eCollection 2025 Sep 9. Mol Ther Nucleic Acids. 2025. PMID: 40673159 Free PMC article.
-
MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs.Cells. 2022 Apr 30;11(9):1509. doi: 10.3390/cells11091509. Cells. 2022. PMID: 35563814 Free PMC article.
References
-
- Stupp R, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. - PubMed
-
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. - PubMed
-
- Lees-Miller SP, et al. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–1185. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
