

Strategies to enable large-scale proteomics for reproducible research
- PMID: 32732981
- PMCID: PMC7393074
- DOI: 10.1038/s41467-020-17641-3
Strategies to enable large-scale proteomics for reproducible research
Abstract
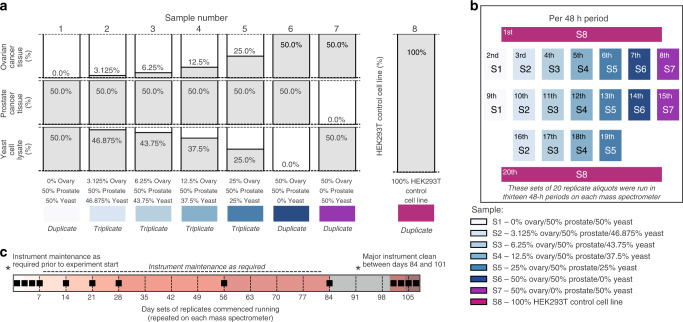
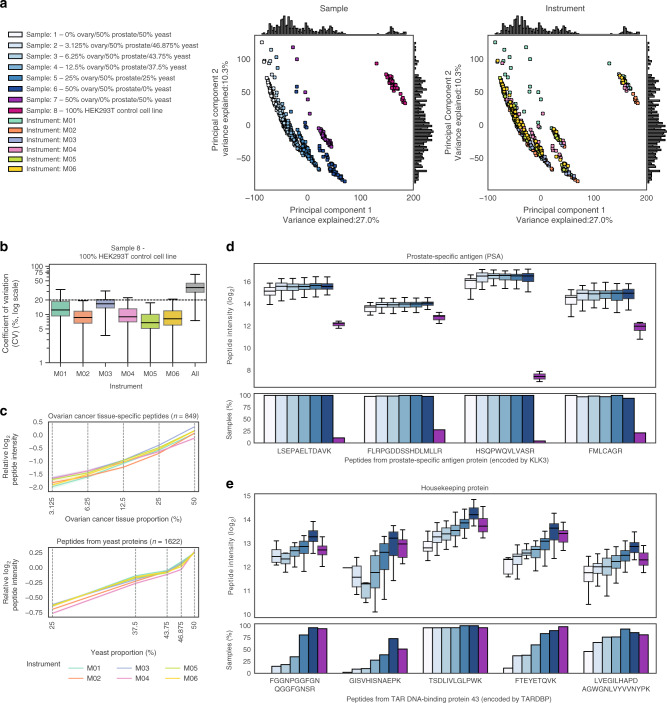
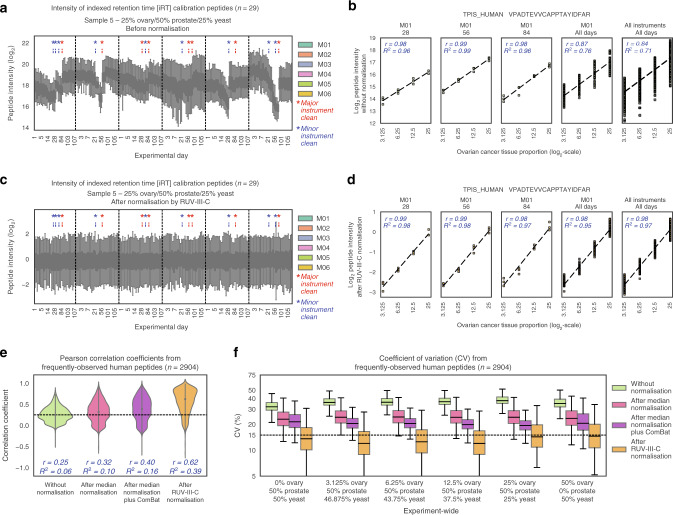
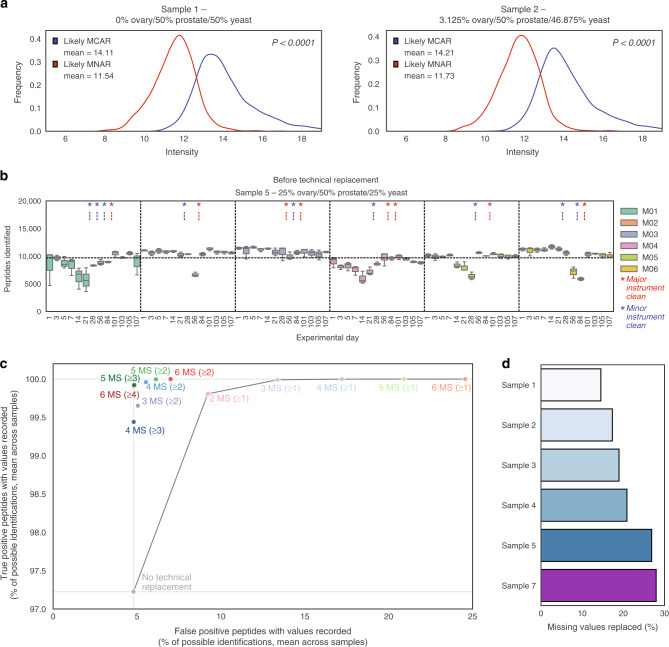
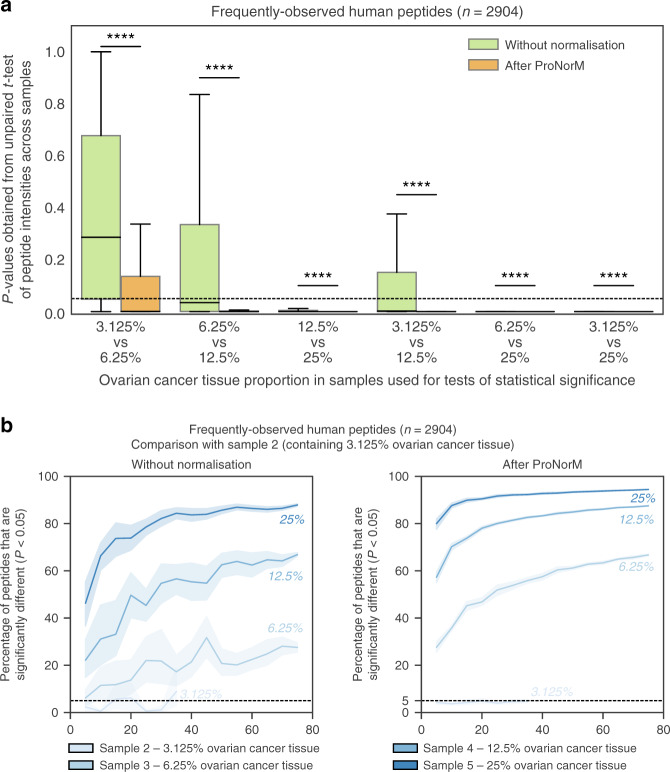
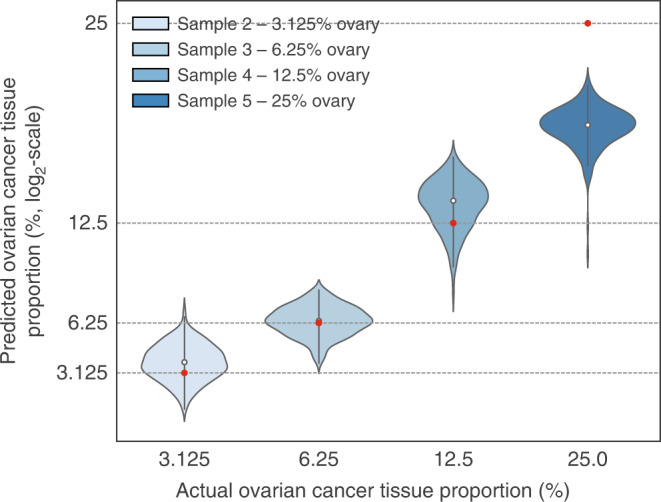
Reproducible research is the bedrock of experimental science. To enable the deployment of large-scale proteomics, we assess the reproducibility of mass spectrometry (MS) over time and across instruments and develop computational methods for improving quantitative accuracy. We perform 1560 data independent acquisition (DIA)-MS runs of eight samples containing known proportions of ovarian and prostate cancer tissue and yeast, or control HEK293T cells. Replicates are run on six mass spectrometers operating continuously with varying maintenance schedules over four months, interspersed with ~5000 other runs. We utilise negative controls and replicates to remove unwanted variation and enhance biological signal, outperforming existing methods. We also design a method for reducing missing values. Integrating these computational modules into a pipeline (ProNorM), we mitigate variation among instruments over time and accurately predict tissue proportions. We demonstrate how to improve the quantitative analysis of large-scale DIA-MS data, providing a pathway toward clinical proteomics.
Conflict of interest statement
K.A. is an employee of SCIEX, which operates in the field covered by the article. R.A. holds shares of Biognosys AG which operates in the field covered by the article. The remaining authors declare no competing interests.
Figures
References
-
- Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389:1134–1150. - PubMed
-
- Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. - PubMed
-
- Tully, B. et al. Addressing the challenges of high-throughput cancer tissue proteomics for clinical application: ProCan®. Proteomics. 10.1002/pmic.201900109 (2019) - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
