

Innate Immune Defense Mechanisms by Myeloid Cells That Hamper Cancer Immunotherapy
- PMID: 32733461
- PMCID: PMC7363805
- DOI: 10.3389/fimmu.2020.01395
Innate Immune Defense Mechanisms by Myeloid Cells That Hamper Cancer Immunotherapy
Abstract
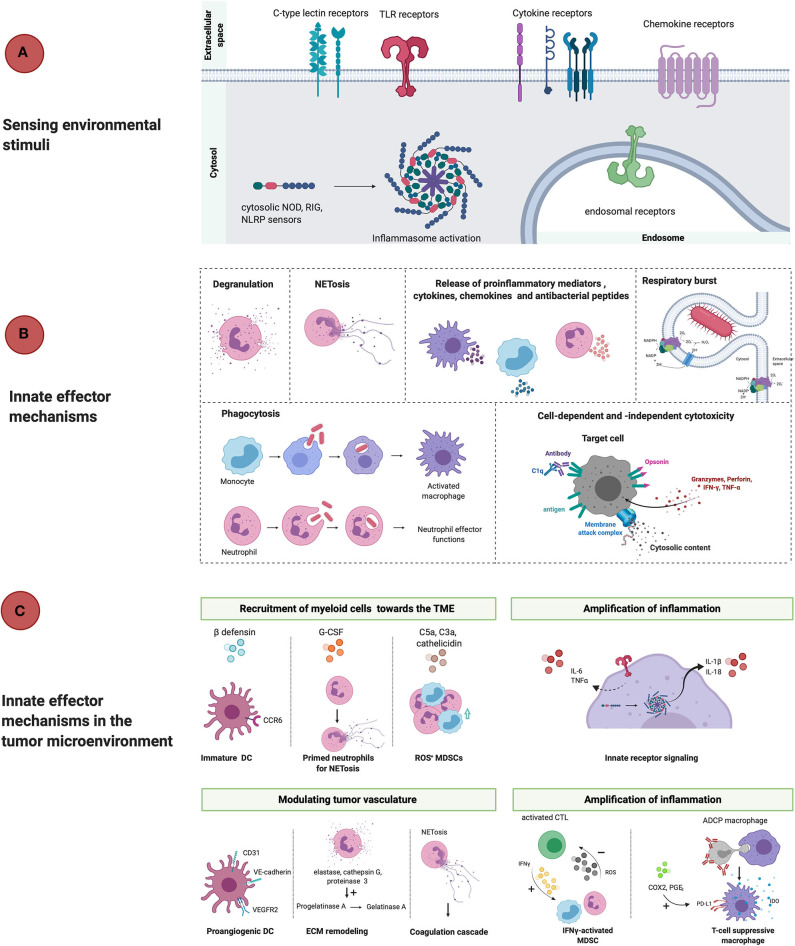
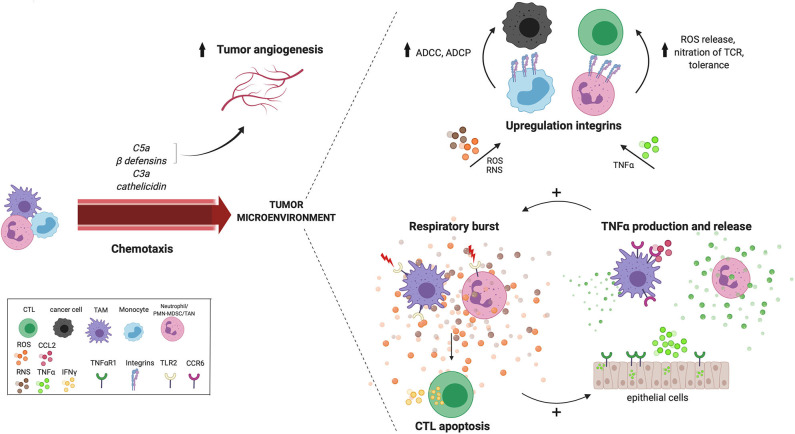
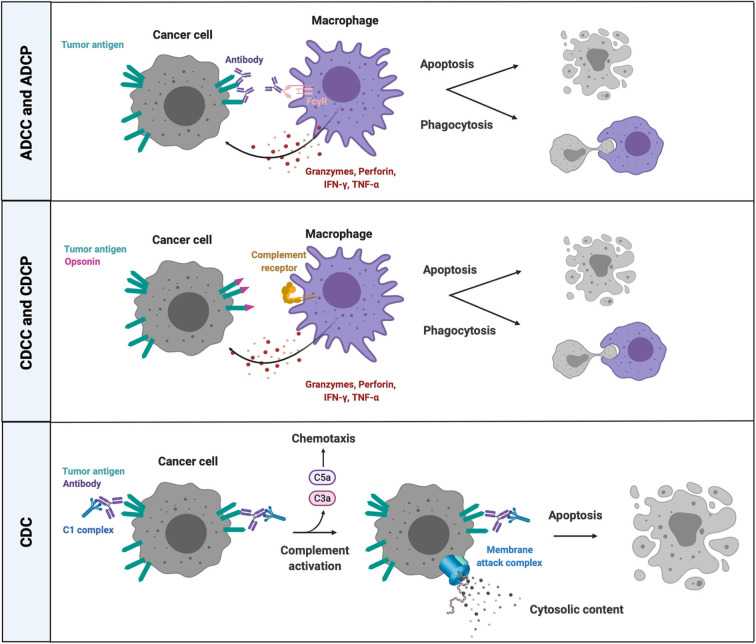
Over the past decade, cancer immunotherapy has been steering immune responses toward cancer cell eradication. However, these immunotherapeutic approaches are hampered by the tumor-promoting nature of myeloid cells, including monocytes, macrophages, and neutrophils. Despite the arsenal of defense strategies against foreign invaders, myeloid cells succumb to the instructions of an established tumor. Interestingly, the most primordial defense responses employed by myeloid cells against pathogens, such as complement activation, antibody-dependent cell cytotoxicity and phagocytosis, actually seem to favor cancer progression. In this review, we discuss how rudimentary defense mechanisms deployed by myeloid cells can promote tumor progression.
Keywords: cancer immunotherapy; immune suppression; immunotherapy resistance; innate immune response; tumor microenvironment; tumor-associated myeloid cells.
Copyright © 2020 Lebegge, Arnouk, Bardet, Kiss, Raes and Van Ginderachter.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous