

Advances in Autosomal Dominant Polycystic Kidney Disease: A Clinical Review
- PMID: 32734239
- PMCID: PMC7380379
- DOI: 10.1016/j.xkme.2019.11.009
Advances in Autosomal Dominant Polycystic Kidney Disease: A Clinical Review
Abstract
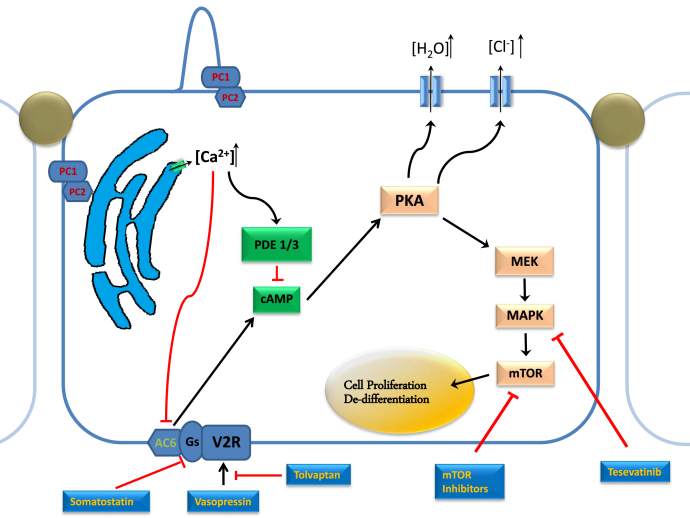
Polycystic kidney disease (PKD) is a multiorgan disorder resulting in fluid-filled cyst formation in the kidneys and other systems. The replacement of kidney parenchyma with an ever-increasing volume of cysts eventually leads to kidney failure. Recently, increased understanding of the pathophysiology of PKD and genetic advances have led to new approaches of treatment targeting physiologic pathways, which has been proven to slow the progression of certain types of the disease. We review the pathophysiologic patterns and recent advances in the clinical pharmacotherapy of autosomal dominant PKD. A multipronged approach with pharmacologic and nonpharmacologic treatments can be successfully used to slow down the rate of progression of autosomal dominant PKD to kidney failure.
Keywords: ADH; ADPKD; Polycystic kidney disease; TKV; autosomal dominant polycystic kidney disease; tolvaptan.
© 2020 The Authors.
Figures
References
-
- Igarashi P., Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13(9):2384–2398. http://www.ncbi.nlm.nih.gov/pubmed/12191984 - PubMed
-
- Helal I. Autosomal dominant polycystic kidney disease: new insights into treatment. Saudi J Kidney Dis Transpl. 2013;24(2):230–234. http://www.ncbi.nlm.nih.gov/pubmed/23538343 - PubMed
-
- Spithoven E.M., Kramer A., Meijer E. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014;86(6):1244–1252. http://www.ncbi.nlm.nih.gov/pubmed/24827775 - PubMed
-
- Collins A.J., Foley R.N., Chavers B. US Renal Data System 2011 Annual Data Report. Am J Kidney Dis. 2012;59(1):A7. http://linkinghub.elsevier.com/retrieve/pii/S027263861101571X - PubMed
-
- Chebib F.T., Torres V.E. Autosomal dominant polycystic kidney disease: core curriculum 2016. Am J Kidney Dis. 2016;67(5):792–810. http://linkinghub.elsevier.com/retrieve/pii/S0272638615012160 - PMC - PubMed
