

Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: natural history and nosological perspectives
- PMID: 32736638
- PMCID: PMC7393722
- DOI: 10.1186/s13023-020-01470-0
Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: natural history and nosological perspectives
Abstract
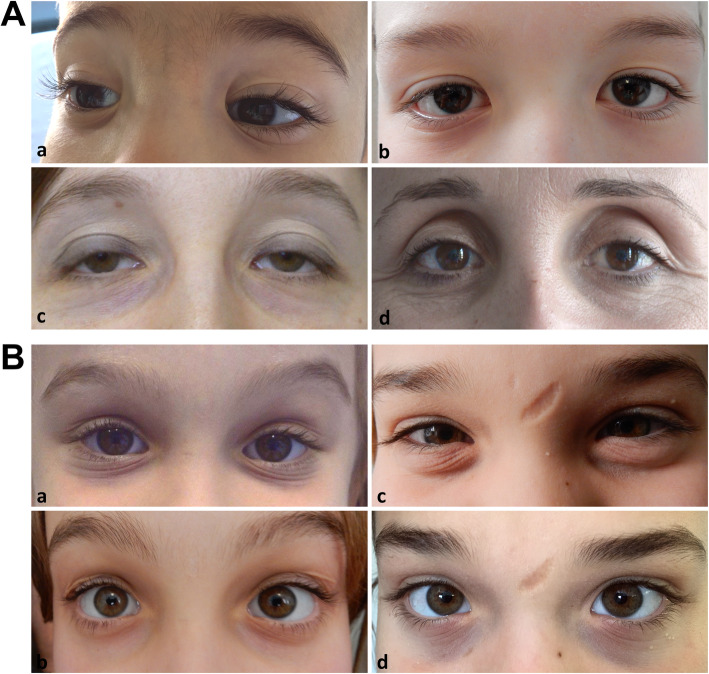
Background: The Ehlers-Danlos syndromes (EDS) are rare connective tissue disorders consisting of 13 subtypes with overlapping features including joint hypermobility, skin and generalized connective tissue fragility. Classical EDS (cEDS) is principally caused by heterozygous COL5A1 or COL5A2 variants and rarely by the COL1A1 p.(Arg312Cys) substitution. Current major criteria are (1) skin hyperextensibility plus atrophic scars and (2) generalized joint hypermobility (gJHM). Minor criteria include additional mucocutaneous signs, epicanthal folds, gJHM complications, and an affected first-degree relative. Minimal criteria prompting molecular testing are major criterion 1 plus either major criterion 2 or 3 minor criteria. In addition to these features, the clinical picture also involves multiple organ systems, but large-scale cohort studies are still missing. This study aimed to investigate the multisystemic involvement and natural history of cEDS through a cross-sectional study on a cohort of 75 molecularly confirmed patients evaluated from 2010 to 2019 in a tertiary referral center. The diagnostic criteria, additional mucocutaneous, osteoarticular, musculoskeletal, cardiovascular, gastrointestinal, uro-gynecological, neuropsychiatric, and atopic issues, and facial/ocular features were ascertained, and feature rates compared by sex and age.
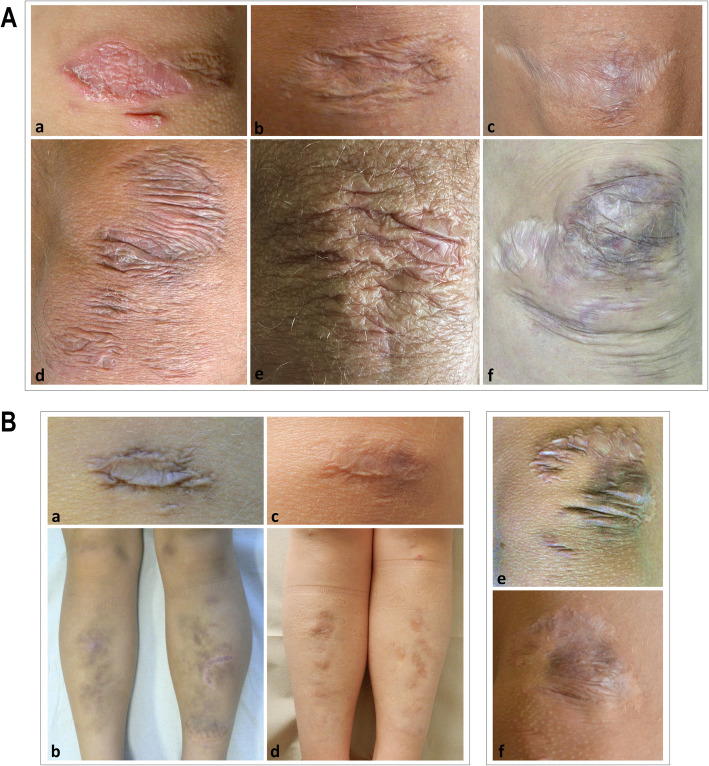
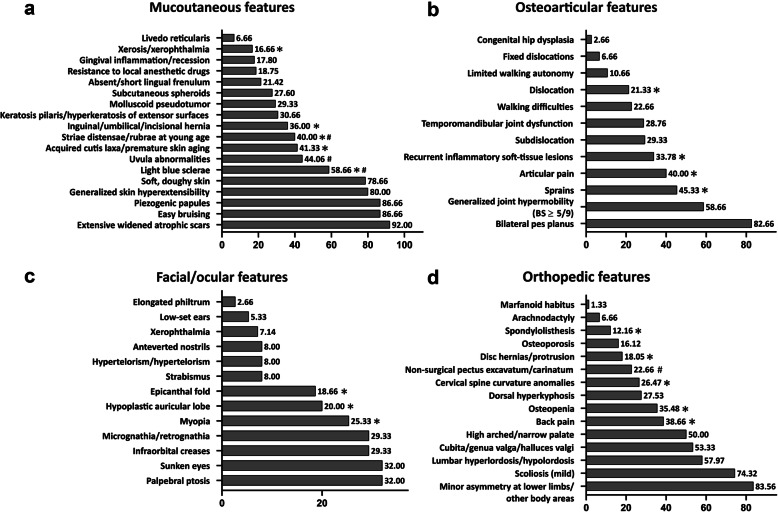
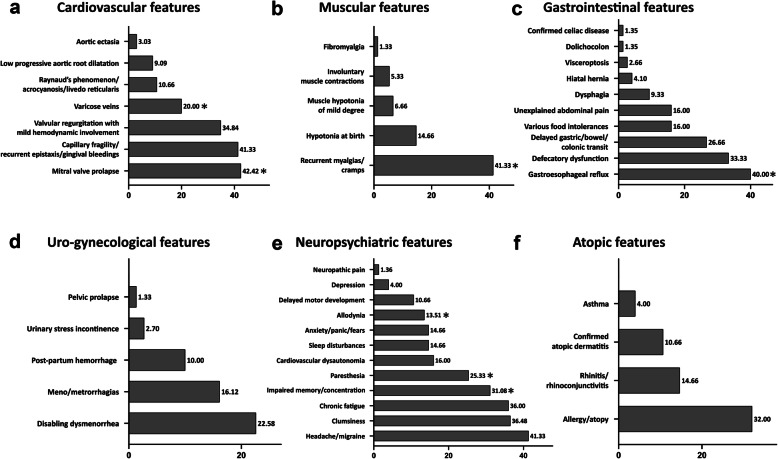
Results: Our study confirms that cEDS is mainly characterized by cutaneous and articular involvement, though none of their hallmarks was represented in all cases and suggests a milder multisystemic involvement and a more favorable natural history compared to other EDS subtypes. Abnormal scarring was the most frequent and characteristic sign, skin hyperextensibility and gJHM were less common, all without any sex and age bias; joint instability complications were more recurrent in adults. Some orthopedic features showed a high prevalence, whereas the other issues related to the investigated organ systems were less recurrent with few exceptions and age-related differences.
Conclusions: Our findings define the diagnostic relevance of cutaneous and articular features and additional clinical signs associated to cEDS. Furthermore, our data suggest an update of the current EDS nosology concerning scarring that should be considered separately from skin hyperextensibility and that the clinical diagnosis of cEDS may be enhanced by the accurate evaluation of orthopedic manifestations at all ages, faciocutaneous indicators in children, and some acquired traits related to joint instability complications, premature skin aging, and patterning of abnormal scarring in older individuals.
Keywords: Atrophic scars; COL1A1; COL5A1; COL5A2; Classical Ehlers-Danlos syndrome; Joint hypermobility; Multisystemic involvement; Natural history; Nosology; Skin hyperextensibility.
Conflict of interest statement
All authors declare that they have no competing interest concerning this work.
Figures
References
-
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am J Med Genet Part C Semin Med Genet. 2017;175:8–26. - PubMed
-
- Malfait F, Coucke P, Symoens S, Loeys B, Nuytinck L, De Paepe A. The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum Mutat. 2005;25:28–37. - PubMed
-
- Symoens S, Syx D, Malfait F, Callewaert B, De Backer J, Vanakker O, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat. 2012;33:1485–1493. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
