

Adoptive T Cell Therapy with IL-12-Preconditioned Low-Avidity T Cells Prevents Exhaustion and Results in Enhanced T Cell Activation, Enhanced Tumor Clearance, and Decreased Risk for Autoimmunity
- PMID: 32737148
- PMCID: PMC8293279
- DOI: 10.4049/jimmunol.2000007
Adoptive T Cell Therapy with IL-12-Preconditioned Low-Avidity T Cells Prevents Exhaustion and Results in Enhanced T Cell Activation, Enhanced Tumor Clearance, and Decreased Risk for Autoimmunity
Abstract
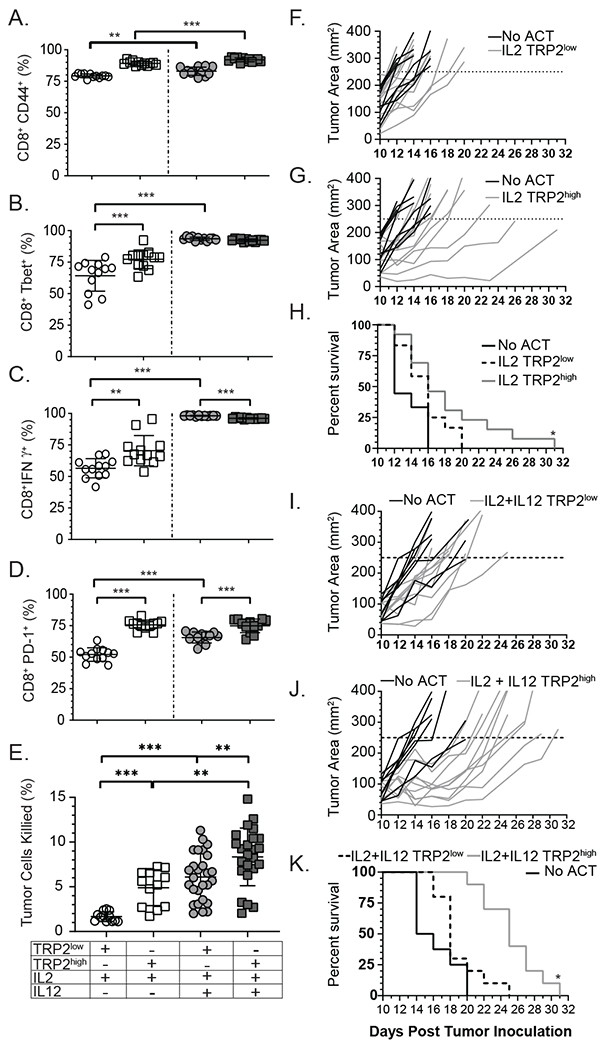
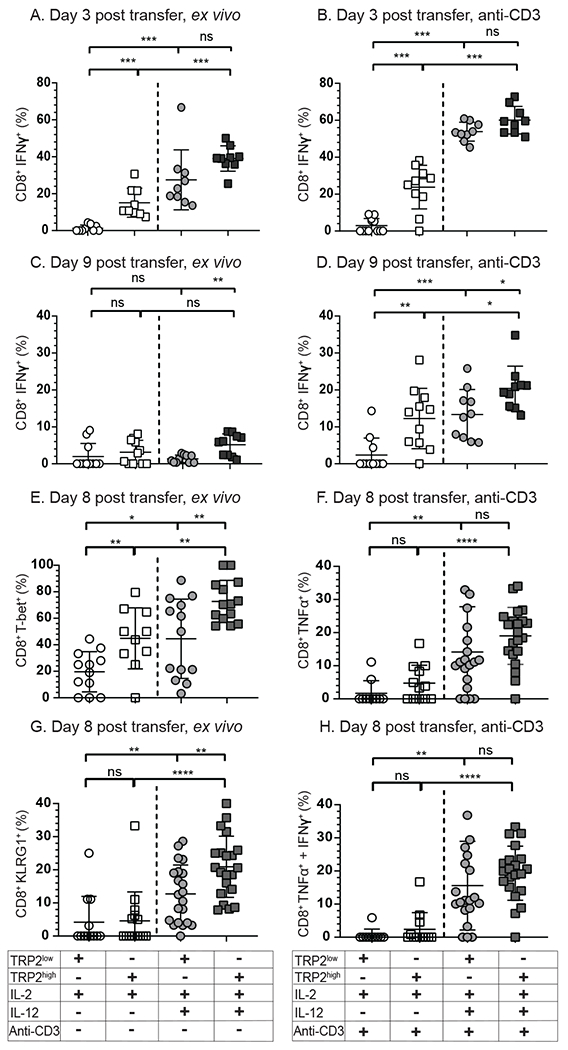
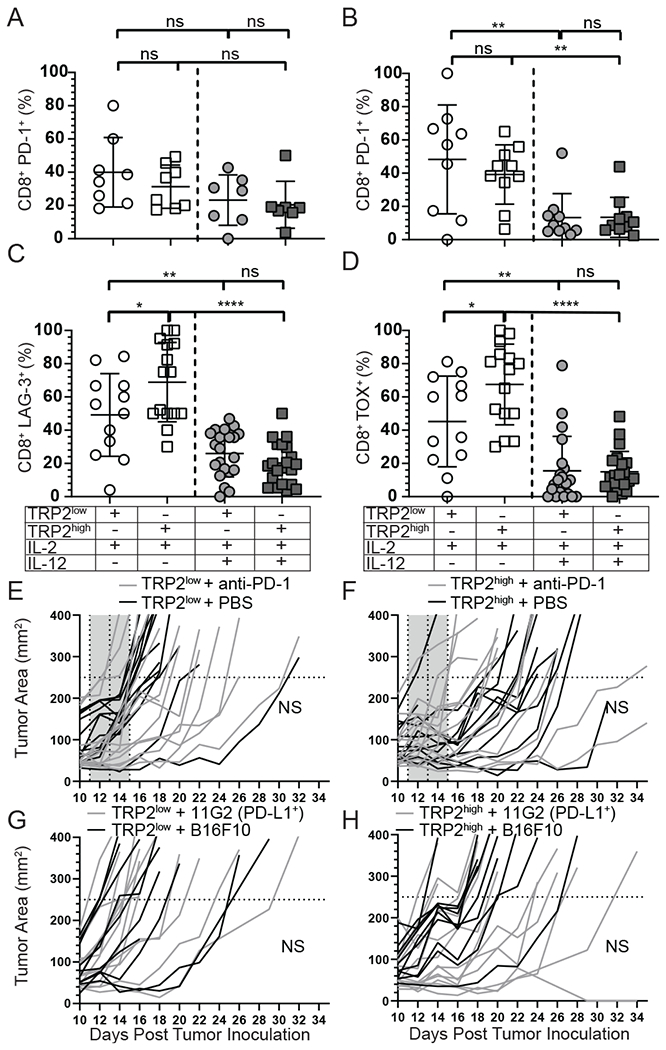
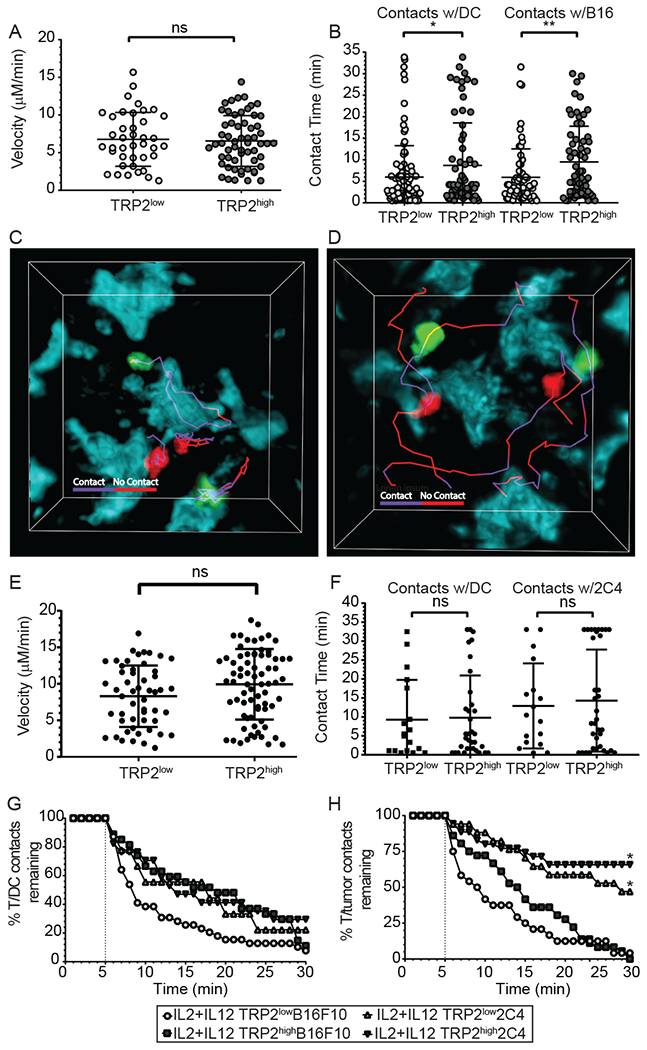
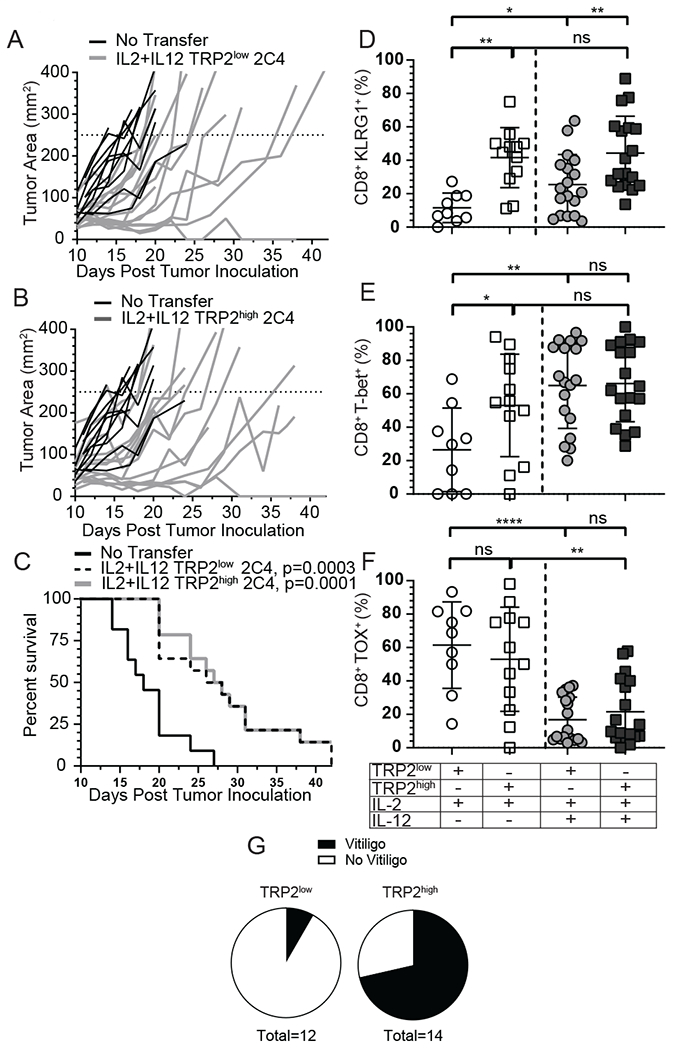
Optimal ex vivo expansion protocols of tumor-specific T cells followed by adoptive cell therapy must yield T cells able to home to tumors and effectively kill them. Our previous study demonstrated ex vivo activation in the presence of IL-12-induced optimal CD8+ T cell expansion and melanoma regression; however, adverse side effects, including autoimmunity, can occur. This may be due to transfer of high-avidity self-specific T cells. In this study, we compared mouse low- and high-avidity T cells targeting the tumor Ag tyrosinase-related protein 2 (TRP2). Not surprisingly, high-avidity T cells provide superior tumor control, yet low-avidity T cells can promote tumor regression. The addition of IL-12 during in vitro expansion boosts low-avidity T cell responsiveness, tumor regression, and prevents T cell exhaustion. In this study, we demonstrate that IL-12-primed T cells are resistant to PD-1/PD-L1-mediated suppression and retain effector function. Importantly, IL-12 preconditioning prevented exhaustion as LAG-3, PD-1, and TOX were decreased while simultaneously increasing KLRG1. Using intravital imaging, we also determined that high-avidity T cells have sustained contacts with intratumoral dendritic cells and tumor targets compared with low-avidity T cells. However, with Ag overexpression, this defect is overcome, and low-avidity T cells control tumor growth. Taken together, these data illustrate that low-avidity T cells can be therapeutically beneficial if cocultured with IL-12 cytokine during in vitro expansion and highly effective in vivo if Ag is not limiting. Clinically, low-avidity T cells provide a safer alternative to high-avidity, TCR-engineered T cells, as IL-12-primed, low-avidity T cells cause less autoimmune vitiligo.
Copyright © 2020 by The American Association of Immunologists, Inc.
Figures
References
-
- Wei SC, Duffy CR, and Allison JP, Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery, 2018. 8(9): p. 1069–1086. - PubMed
-
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Drilon A, Wolchok JD, Carvajal RD, McHenry MB, Hosein F, Harbison CT, Grosso JF, and Sznol M, Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol, 2019. - PMC - PubMed
-
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, and Ribas A, PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 2014. 515(7528): p. 568–571. - PMC - PubMed
-
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, and Wherry EJ, Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science, 2016. 354(6316): p. 1160–1165. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
