

Symmetric neural progenitor divisions require chromatin-mediated homologous recombination DNA repair by Ino80
- PMID: 32737294
- PMCID: PMC7395731
- DOI: 10.1038/s41467-020-17551-4
Symmetric neural progenitor divisions require chromatin-mediated homologous recombination DNA repair by Ino80
Abstract
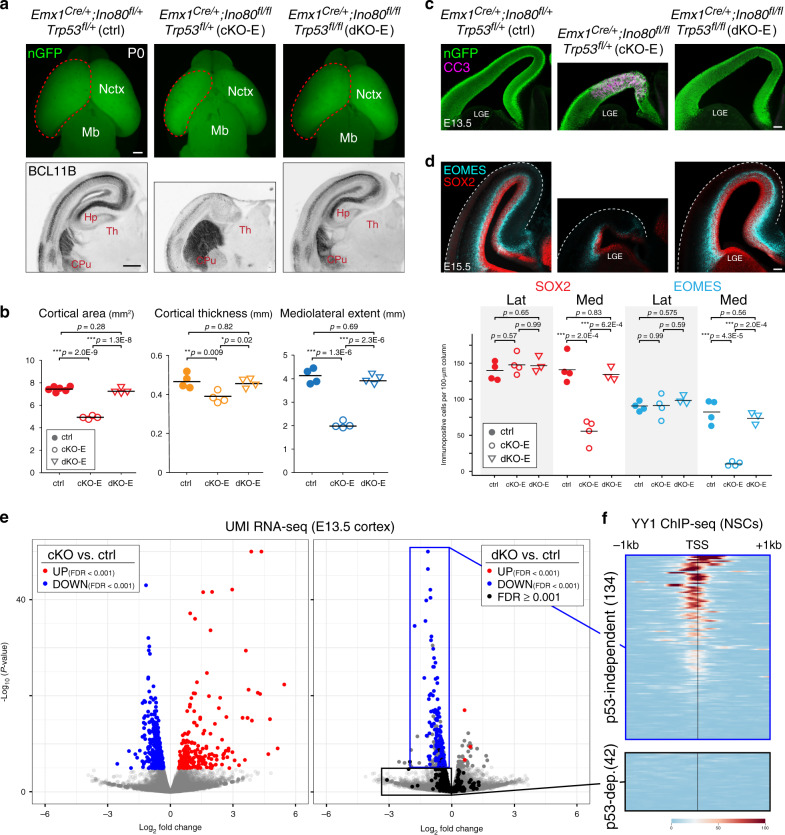
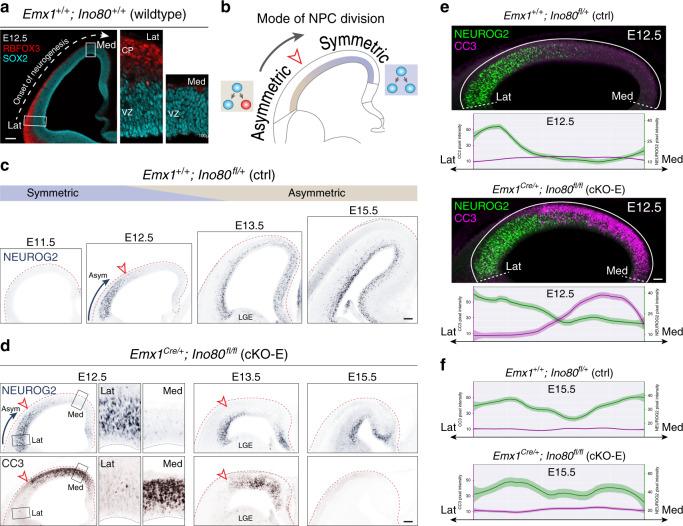
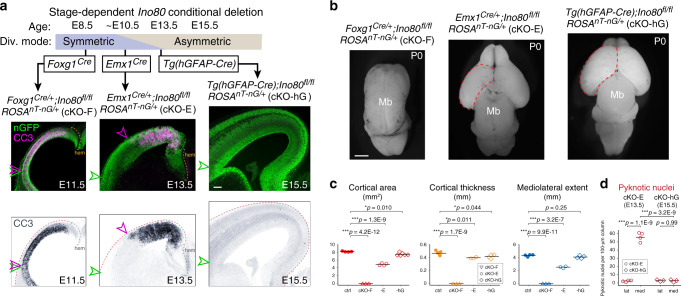
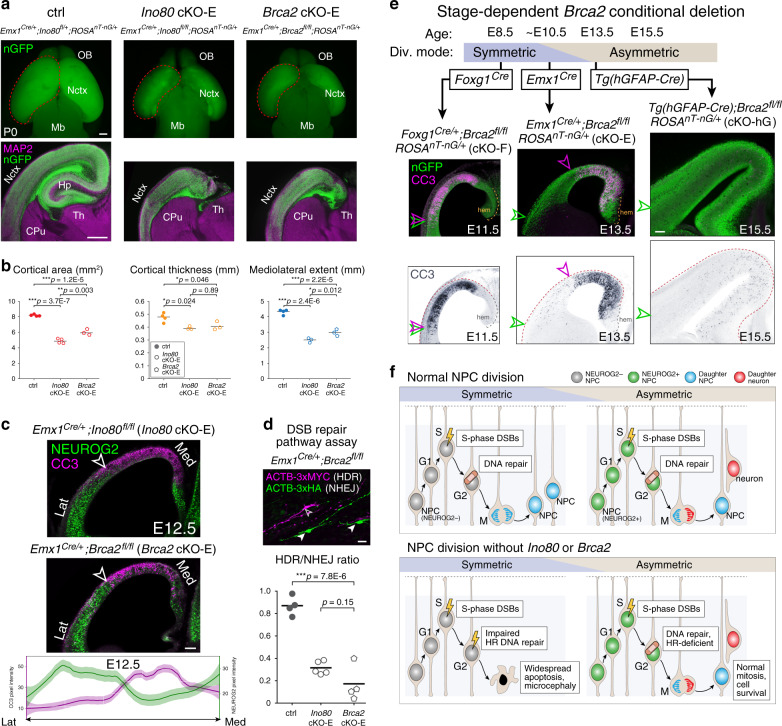
Chromatin regulates spatiotemporal gene expression during neurodevelopment, but it also mediates DNA damage repair essential to proliferating neural progenitor cells (NPCs). Here, we uncover molecularly dissociable roles for nucleosome remodeler Ino80 in chromatin-mediated transcriptional regulation and genome maintenance in corticogenesis. We find that conditional Ino80 deletion from cortical NPCs impairs DNA double-strand break (DSB) repair, triggering p53-dependent apoptosis and microcephaly. Using an in vivo DSB repair pathway assay, we find that Ino80 is selectively required for homologous recombination (HR) DNA repair, which is mechanistically distinct from Ino80 function in YY1-associated transcription. Unexpectedly, sensitivity to loss of Ino80-mediated HR is dependent on NPC division mode: Ino80 deletion leads to unrepaired DNA breaks and apoptosis in symmetric NPC-NPC divisions, but not in asymmetric neurogenic divisions. This division mode dependence is phenocopied following conditional deletion of HR gene Brca2. Thus, distinct modes of NPC division have divergent requirements for Ino80-dependent HR DNA repair.
Conflict of interest statement
The authors declare no competing interests.
Figures
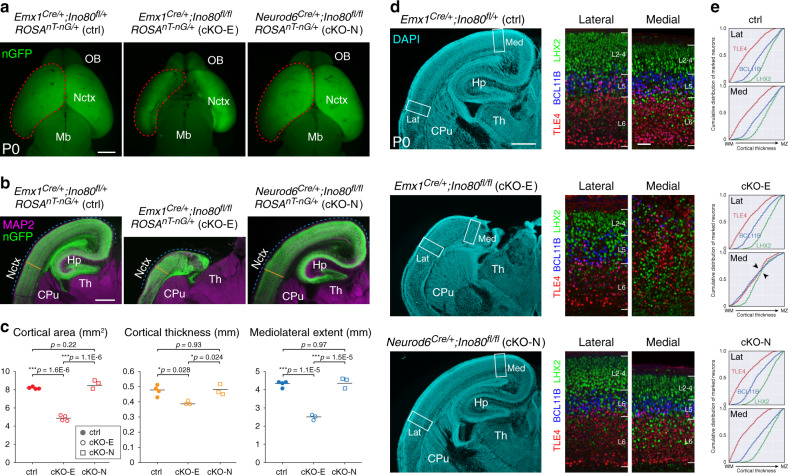
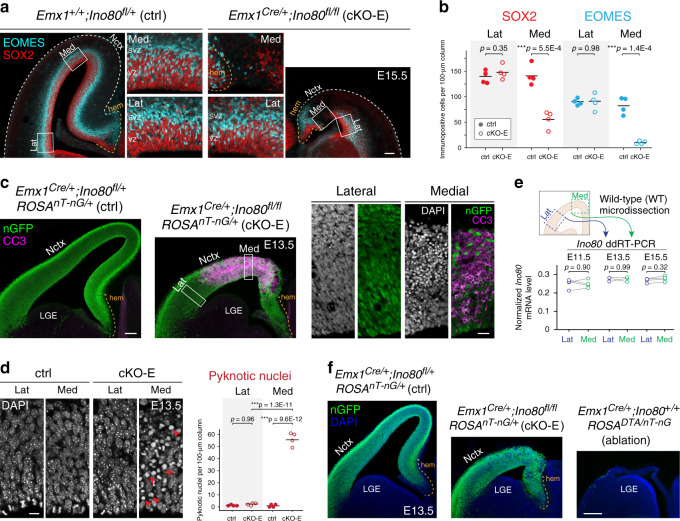
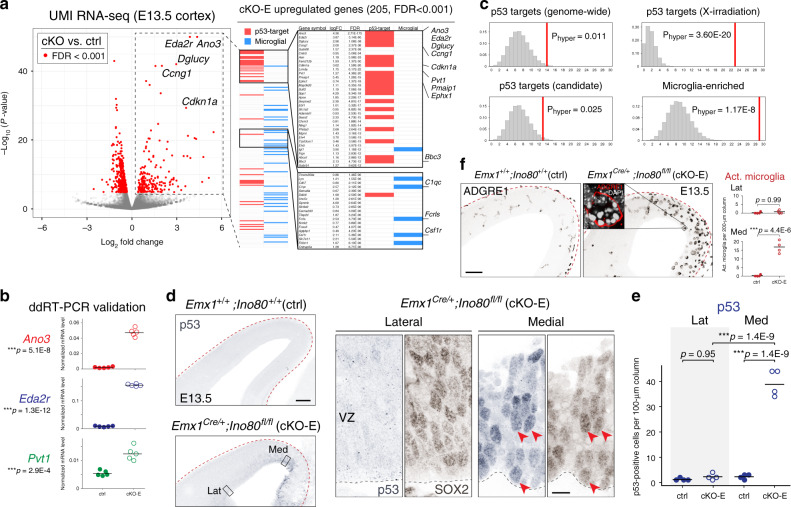
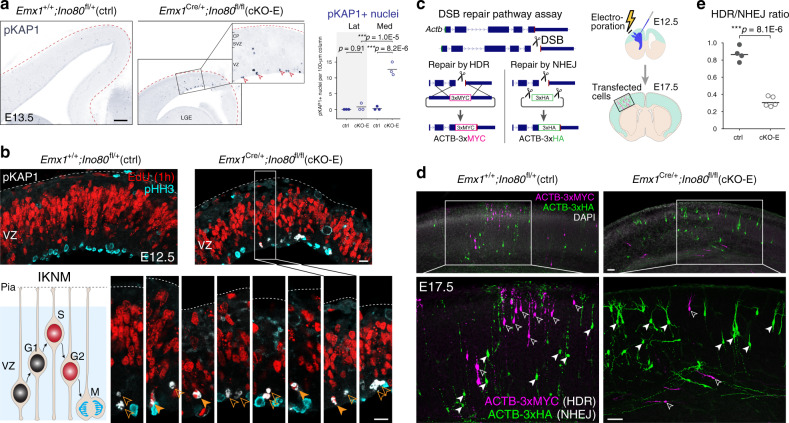
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
