

Assessing non-Mendelian inheritance in inherited axonopathies
- PMID: 32741968
- PMCID: PMC7710562
- DOI: 10.1038/s41436-020-0924-0
Assessing non-Mendelian inheritance in inherited axonopathies
Abstract
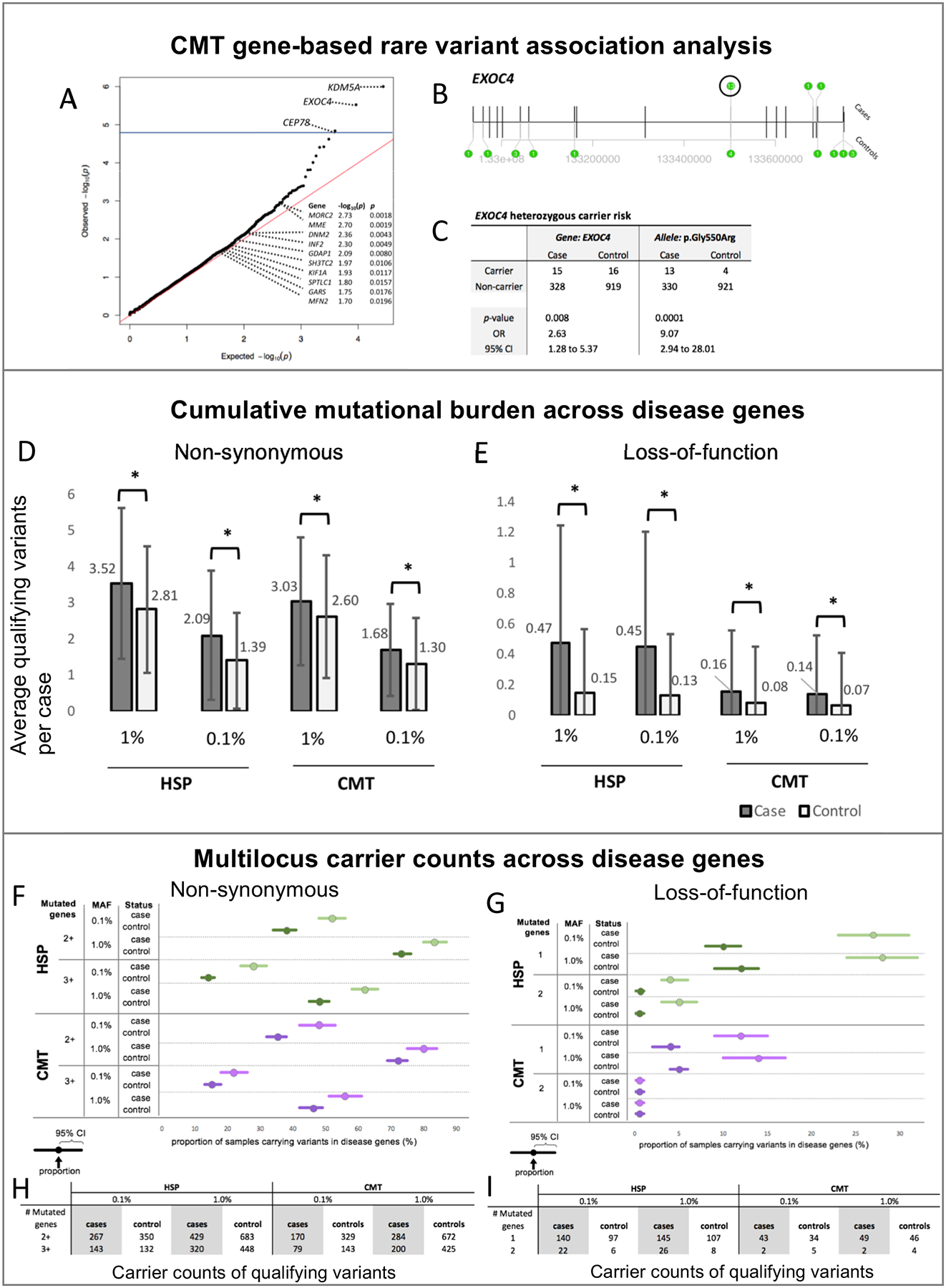
Purpose: Inherited axonopathies (IA) are rare, clinically and genetically heterogeneous diseases that lead to length-dependent degeneration of the long axons in central (hereditary spastic paraplegia [HSP]) and peripheral (Charcot-Marie-Tooth type 2 [CMT2]) nervous systems. Mendelian high-penetrance alleles in over 100 different genes have been shown to cause IA; however, about 50% of IA cases do not receive a genetic diagnosis. A more comprehensive spectrum of causative genes and alleles is warranted, including causative and risk alleles, as well as oligogenic multilocus inheritance.
Methods: Through international collaboration, IA exome studies are beginning to be sufficiently powered to perform a pilot rare variant burden analysis. After extensive quality control, our cohort contained 343 CMT cases, 515 HSP cases, and 935 non-neurological controls. We assessed the cumulative mutational burden across disease genes, explored the evidence for multilocus inheritance, and performed an exome-wide rare variant burden analysis.
Results: We replicated the previously described mutational burden in a much larger cohort of CMT cases, and observed the same effect in HSP cases. We identified a preliminary risk allele for CMT in the EXOC4 gene (p value= 6.9 × 10-6, odds ratio [OR] = 2.1) and explored the possibility of multilocus inheritance in IA.
Conclusion: Our results support the continuing emergence of complex inheritance mechanisms in historically Mendelian disorders.
Keywords: Charcot–Marie–Tooth disease; hereditary spastic paraplegia; inherited axonopathy; mutational burden; oligogenic inheritance.
Conflict of interest statement
Disclosure: The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
