

This is a preprint.
Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with an endemic human CoV
- PMID: 32743570
- PMCID: PMC7386487
- DOI: 10.1101/2020.07.27.222943
Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with an endemic human CoV
Update in
-
Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with endemic human coronaviruses.Cell Rep Med. 2021 Jan 19;2(1):100189. doi: 10.1016/j.xcrm.2020.100189. Cell Rep Med. 2021. PMID: 33495758 Free PMC article.
Abstract
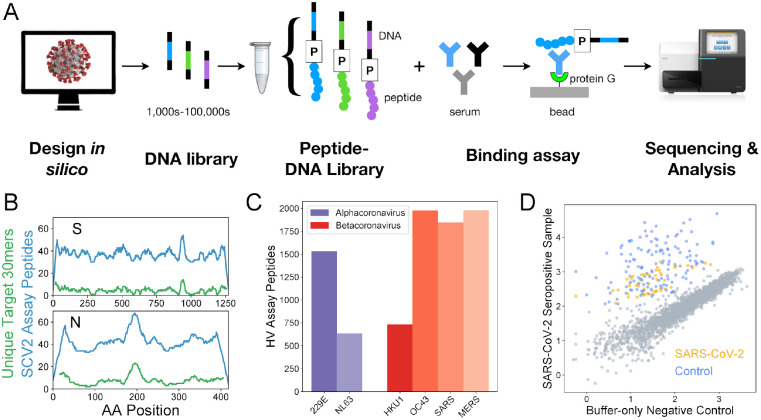
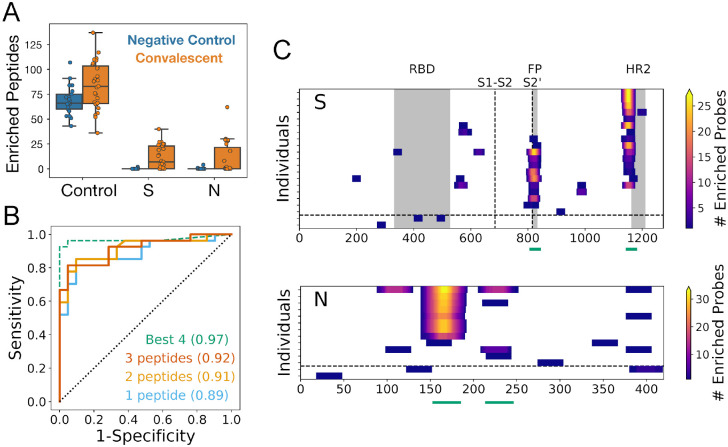
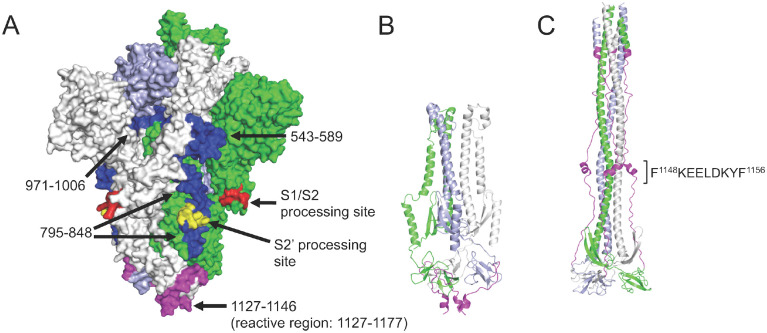
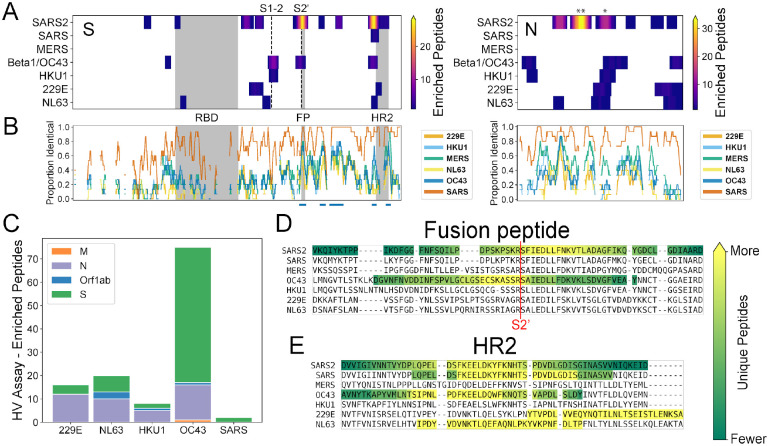
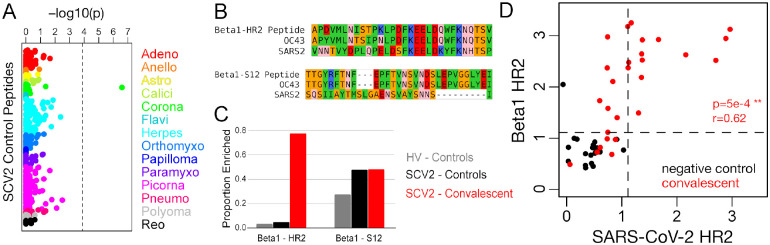
A high-resolution understanding of the antibody response to SARS-CoV-2 is important for the design of effective diagnostics, vaccines and therapeutics. However, SARS-CoV-2 antibody epitopes remain largely uncharacterized, and it is unknown whether and how the response may cross-react with related viruses. Here, we use a multiplexed peptide assay ('PepSeq') to generate an epitope-resolved view of reactivity across all human coronaviruses. PepSeq accurately detects SARS-CoV-2 exposure and resolves epitopes across the Spike and Nucleocapsid proteins. Two of these represent recurrent reactivities to conserved, functionally-important sites in the Spike S2 subunit, regions that we show are also targeted for the endemic coronaviruses in pre-pandemic controls. At one of these sites, we demonstrate that the SARS-CoV-2 response strongly and recurrently cross-reacts with the endemic virus hCoV-OC43. Our analyses reveal new diagnostic and therapeutic targets, including a site at which SARS-CoV-2 may recruit common pre-existing antibodies and with the potential for broadly-neutralizing responses.
Figures
References
-
- Chang C.-K., Hsu Y.-L., Chang Y.-H., Chao F.-A., Wu M.-C., Huang Y.-S., Hu C.-K., and Huang T.-H. (2009). Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J. Virol. 83, 2255–2264. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous