

Pompe disease: pathogenesis, molecular genetics and diagnosis
- PMID: 32745073
- PMCID: PMC7467391
- DOI: 10.18632/aging.103794
Pompe disease: pathogenesis, molecular genetics and diagnosis
Abstract
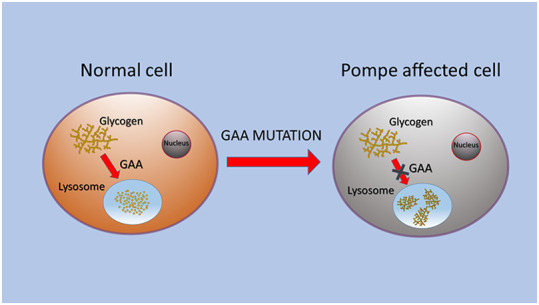
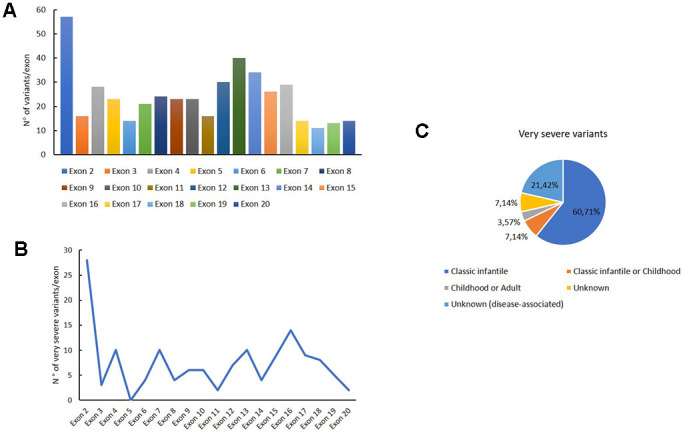
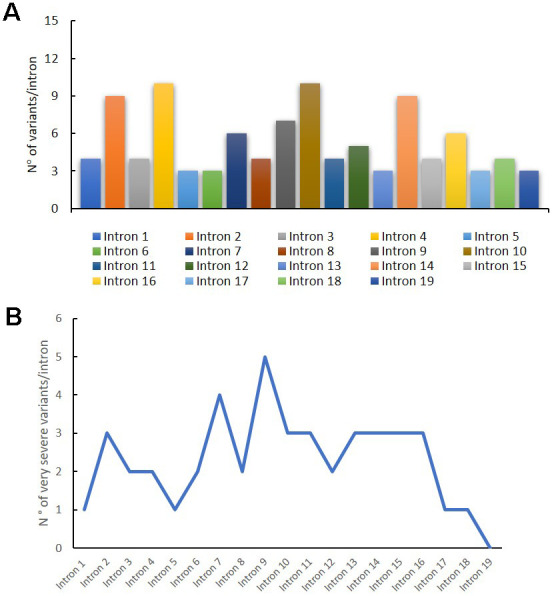
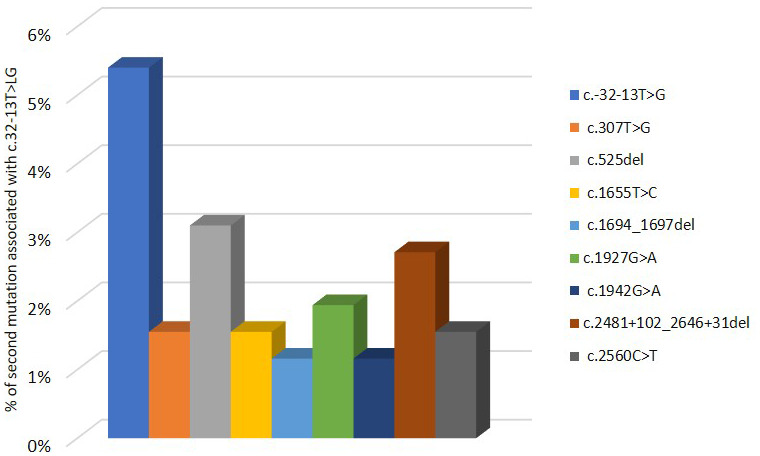
Pompe disease (PD) is a rare autosomal recessive disorder caused by mutations in the GAA gene, localized on chromosome 17 and encoding for acid alpha-1,4-glucosidase (GAA). Currently, more than 560 mutations spread throughout GAA gene have been reported. GAA catalyzes the hydrolysis of α-1,4 and α-1,6-glucosidic bonds of glycogen and its deficiency leads to lysosomal storage of glycogen in several tissues, particularly in muscle. PD is a chronic and progressive pathology usually characterized by limb-girdle muscle weakness and respiratory failure. PD is classified as infantile and childhood/adult forms. PD patients exhibit a multisystemic manifestation that depends on age of onset.Early diagnosis is essential to prevent or reduce the irreversible organ damage associated with PD progression. Here, we make an overview of PD focusing on pathogenesis, clinical phenotypes, molecular genetics, diagnosis, therapies, autophagy and the role of miRNAs as potential biomarkers for PD.
Keywords: GAA; Pompe disease; acid alpha-1,4-glucosidase; glycogen; lysosomal storage disorder.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
