

Extended interaction networks with HCV protease NS3-4A substrates explain the lack of adaptive capability against protease inhibitors
- PMID: 32747444
- PMCID: PMC7535904
- DOI: 10.1074/jbc.RA120.013898
Extended interaction networks with HCV protease NS3-4A substrates explain the lack of adaptive capability against protease inhibitors
Abstract
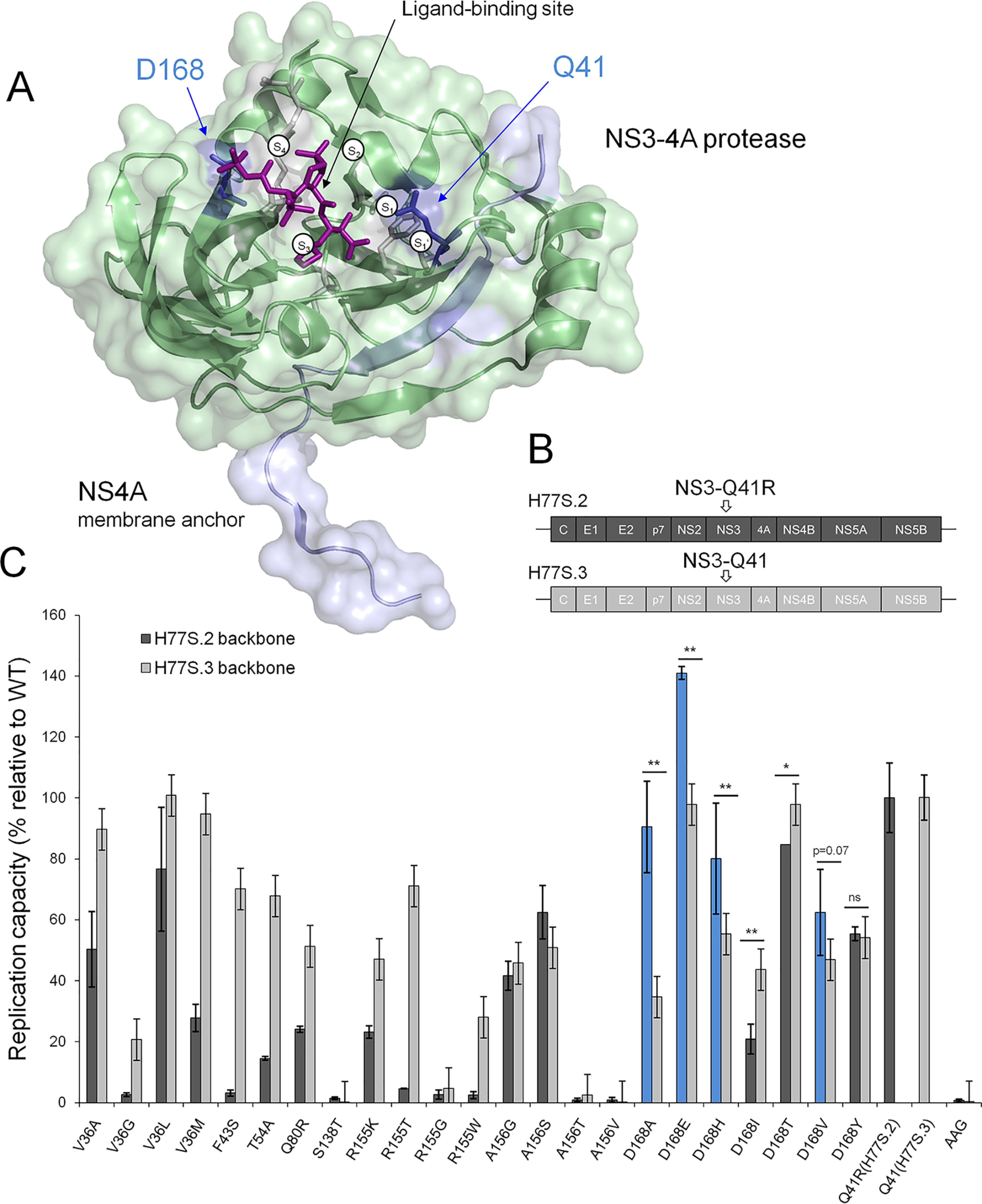
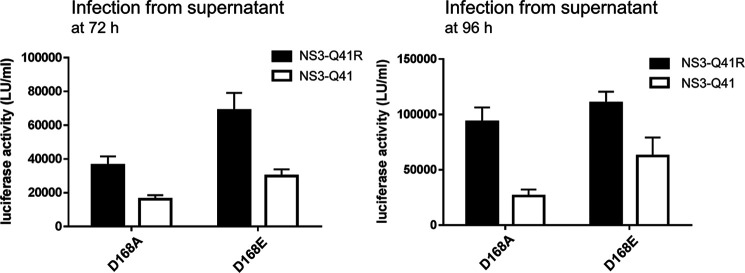
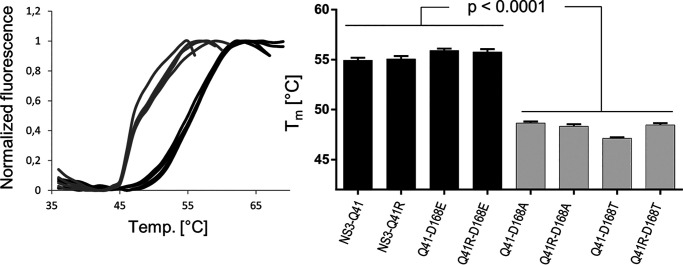
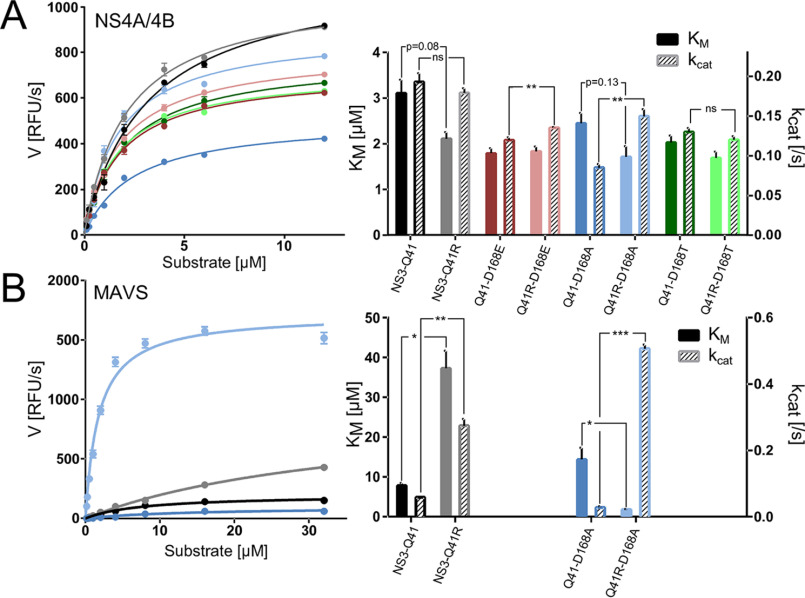
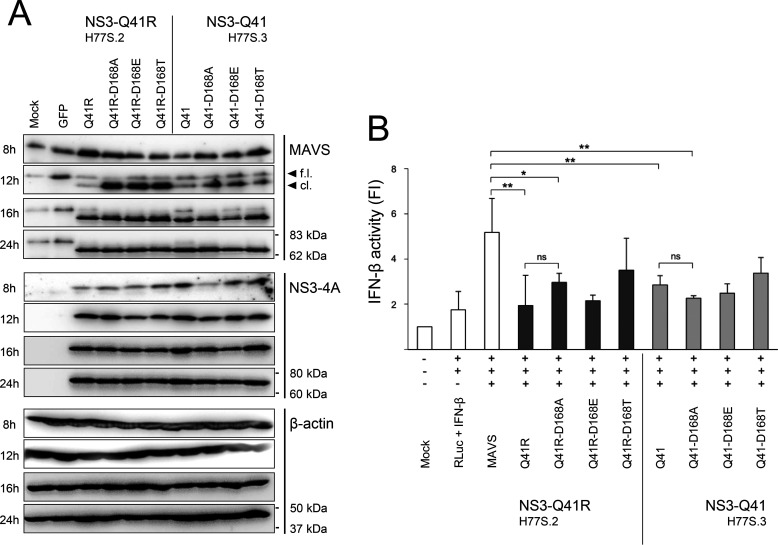
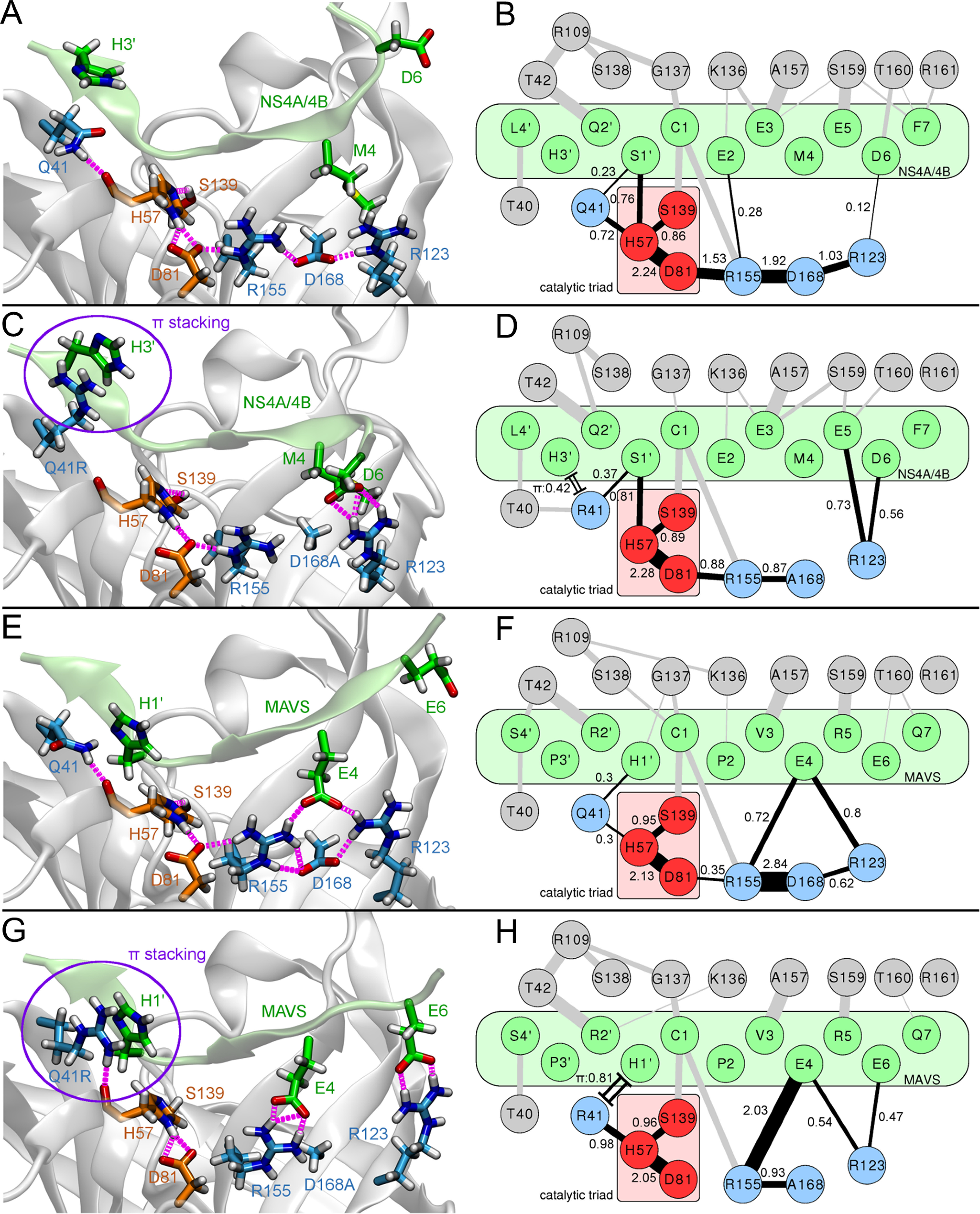
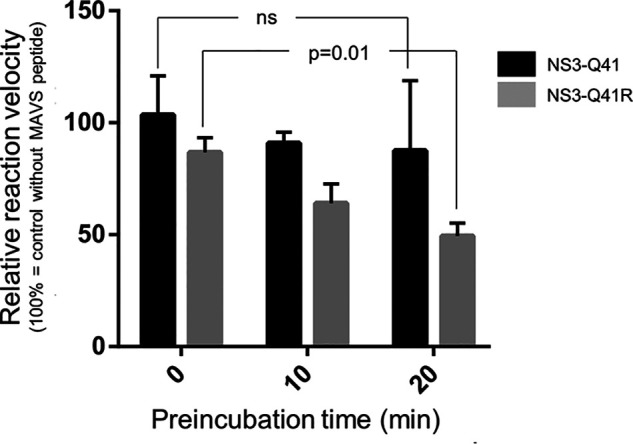
Inhibitors against the NS3-4A protease of hepatitis C virus (HCV) have proven to be useful drugs in the treatment of HCV infection. Although variants have been identified with mutations that confer resistance to these inhibitors, the mutations do not restore replicative fitness and no secondary mutations that rescue fitness have been found. To gain insight into the molecular mechanisms underlying the lack of fitness compensation, we screened known resistance mutations in infectious HCV cell culture with different genomic backgrounds. We observed that the Q41R mutation of NS3-4A efficiently rescues the replicative fitness in cell culture for virus variants containing mutations at NS3-Asp168 To understand how the Q41R mutation rescues activity, we performed protease activity assays complemented by molecular dynamics simulations, which showed that protease-peptide interactions far outside the targeted peptide cleavage sites mediate substrate recognition by NS3-4A and support protease cleavage kinetics. These interactions shed new light on the mechanisms by which NS3-4A cleaves its substrates, viral polyproteins and a prime cellular antiviral adaptor protein, the mitochondrial antiviral signaling protein MAVS. Peptide binding is mediated by an extended hydrogen-bond network in NS3-4A that was effectively optimized for protease-MAVS binding in Asp168 variants with rescued replicative fitness from NS3-Q41R. In the protease harboring NS3-Q41R, the N-terminal cleavage products of MAVS retained high affinity to the active site, rendering the protease susceptible for potential product inhibition. Our findings reveal delicately balanced protease-peptide interactions in viral replication and immune escape that likely restrict the protease adaptive capability and narrow the virus evolutionary space.
Keywords: adaptation; drug resistance; evolution; hepatitis C virus (HCV); mitochondrial antiviral signaling protein (MAVS); molecular adaptation; molecular biology; molecular dynamics; protease inhibitor; replicative fitness; resistance mutation; serine protease (NS3-4A); structure constraints.
© 2020 Dultz et al.
Conflict of interest statement
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Sheldon J., Beach N. M., Moreno E., Gallego I., Piñeiro D., Martínez-Salas E., Gregori J., Quer J., Esteban J. I., Rice C. M., Domingo E., and Perales C. (2014) Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J. Virol. 88, 12098–12111 10.1128/JVI.01860-14 - DOI - PMC - PubMed
-
- Doncheva N. T., Domingues F. S., McGivern D. R., Shimakami T., Zeuzem S., Lengauer T., Lange C. M., Albrecht M., and Welsch C. (2019) Near-neighbor interactions in the NS3-4A protease of HCV impact replicative fitness of drug-resistant viral variants. J. Mol. Biol. 431, 2354–2368 10.1016/j.jmb.2019.04.034 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
