

MethylResolver-a method for deconvoluting bulk DNA methylation profiles into known and unknown cell contents
- PMID: 32747663
- PMCID: PMC7400544
- DOI: 10.1038/s42003-020-01146-2
MethylResolver-a method for deconvoluting bulk DNA methylation profiles into known and unknown cell contents
Abstract
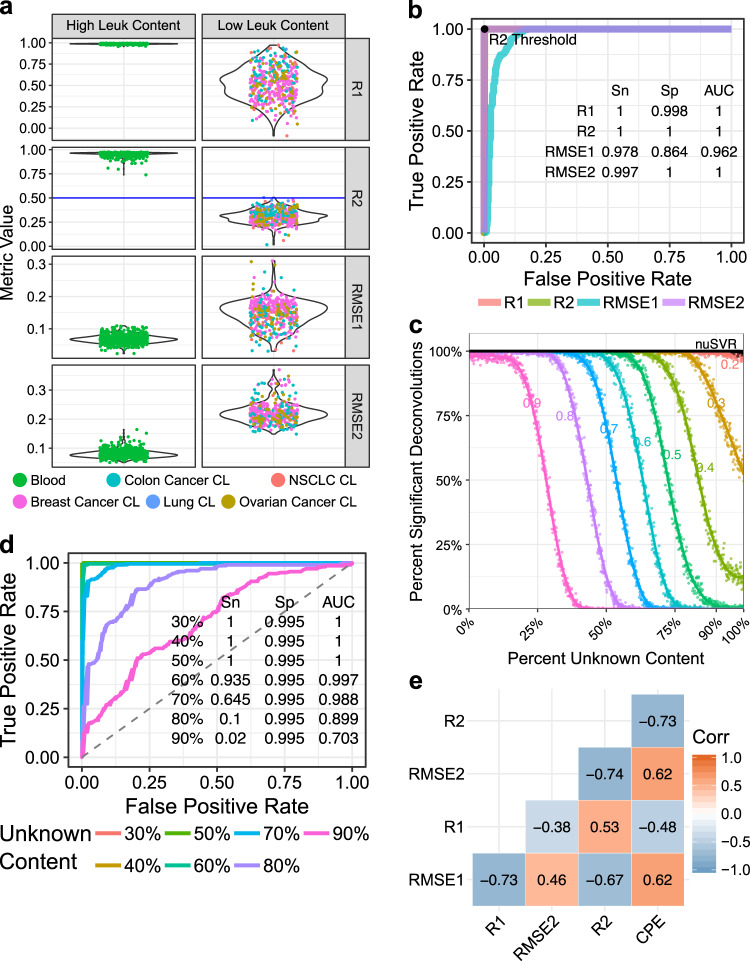
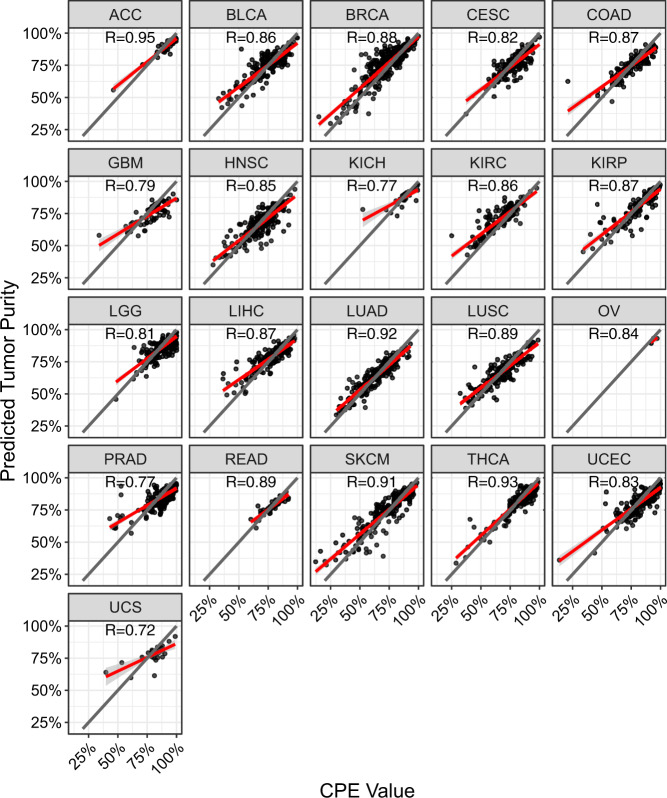
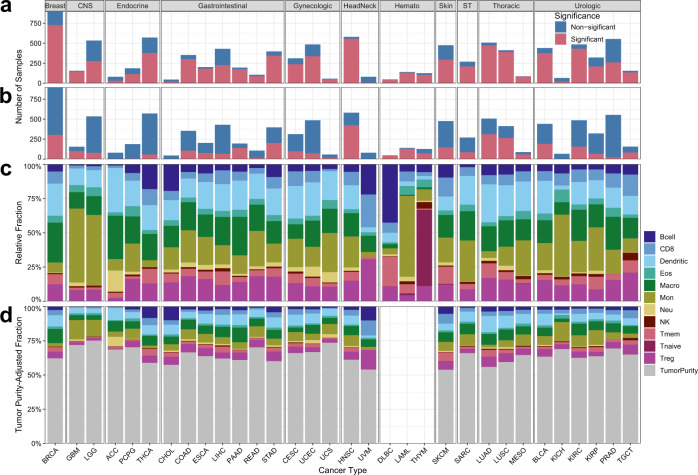
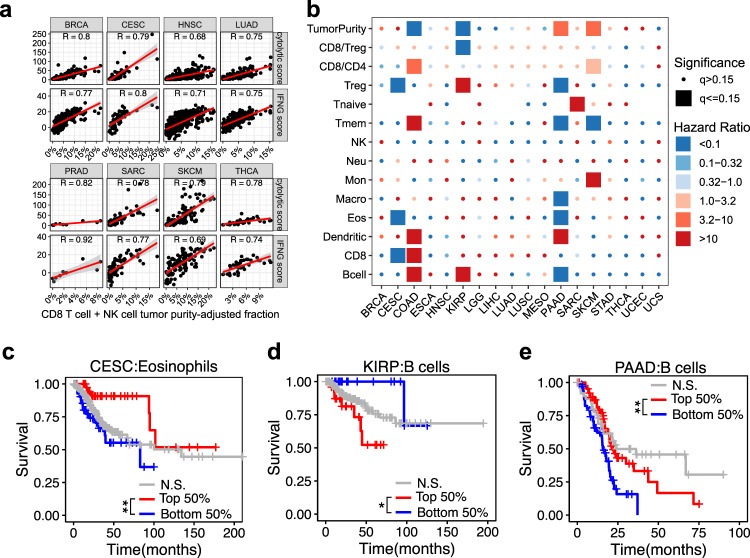
Bulk tissue DNA methylation profiling has been used to examine epigenetic mechanisms and biomarkers of complex diseases such as cancer. However, heterogeneity of cellular content in tissues complicates result interpretation and utility. In silico deconvolution of cellular fractions from bulk tissue data offers a fast and inexpensive alternative to experimentally measuring such fractions. In this study, we report the design, implementation, and benchmarking of MethylResolver, a Least Trimmed Squares regression-based method for inferring leukocyte subset fractions from methylation profiles of tumor admixtures. Compared to previous approaches MethylResolver is more accurate as unknown cellular content in the mixture increases and is able to resolve tumor purity-scaled immune cell-type fractions without a cancer-specific signature. We also present a pan-cancer deconvolution of TCGA, recapitulating that high eosinophil fraction predicts improved cervical carcinoma survival and identifying elevated B cell fraction as a previously unreported predictor of poor survival for papillary renal cell carcinoma.
Conflict of interest statement
K.W. is an employee and shareholder of Bristol-Myers Squibb.
Figures

References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
