

Rational discovery of molecular glue degraders via scalable chemical profiling
- PMID: 32747809
- PMCID: PMC7116640
- DOI: 10.1038/s41589-020-0594-x
Rational discovery of molecular glue degraders via scalable chemical profiling
Erratum in
-
Publisher Correction: Rational discovery of molecular glue degraders via scalable chemical profiling.Nat Chem Biol. 2021 Mar;17(3):361. doi: 10.1038/s41589-021-00735-4. Nat Chem Biol. 2021. PMID: 33452497 No abstract available.
Abstract
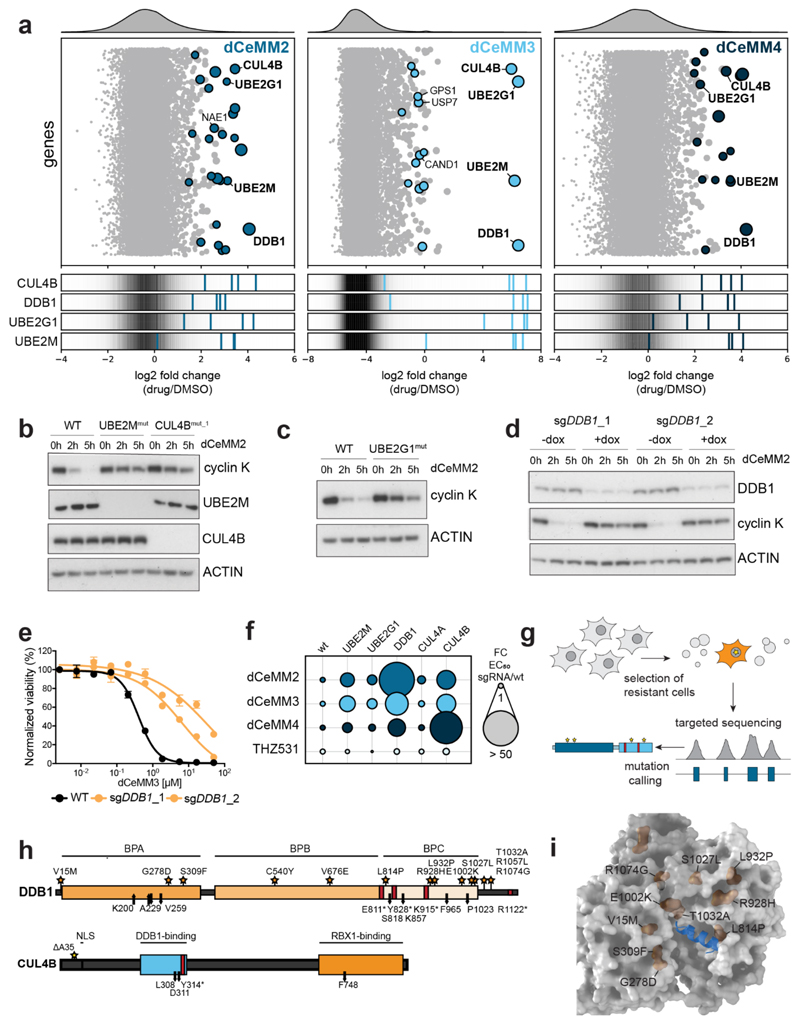
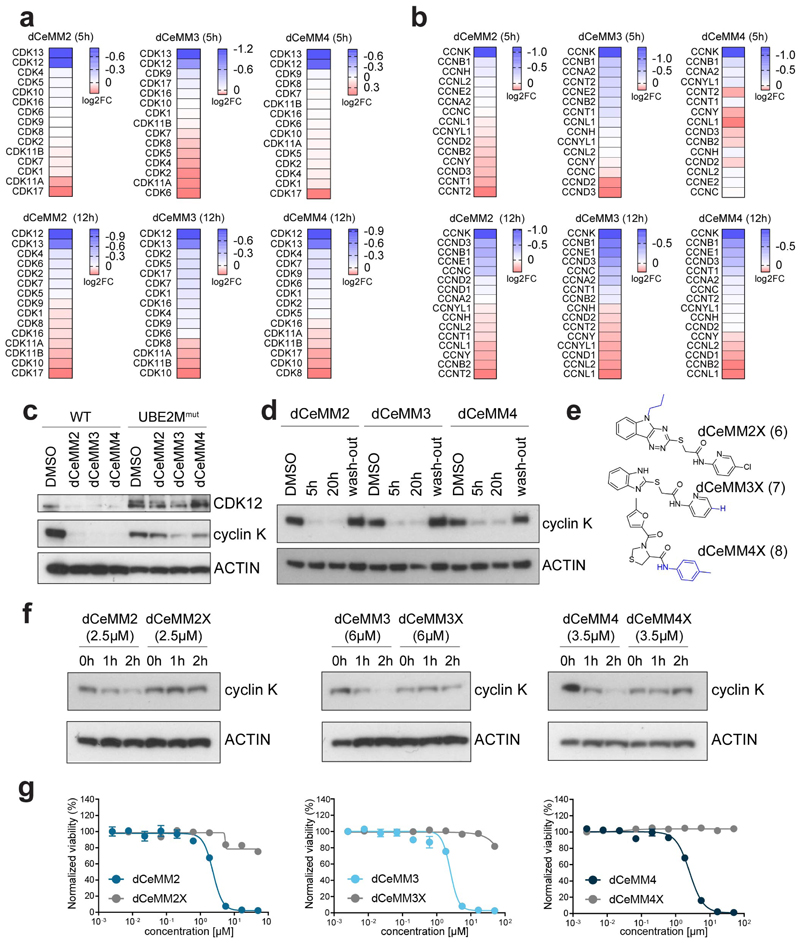
Targeted protein degradation is a new therapeutic modality based on drugs that destabilize proteins by inducing their proximity to E3 ubiquitin ligases. Of particular interest are molecular glues that can degrade otherwise unligandable proteins by orchestrating direct interactions between target and ligase. However, their discovery has so far been serendipitous, thus hampering broad translational efforts. Here, we describe a scalable strategy toward glue degrader discovery that is based on chemical screening in hyponeddylated cells coupled to a multi-omics target deconvolution campaign. This approach led us to identify compounds that induce ubiquitination and degradation of cyclin K by prompting an interaction of CDK12-cyclin K with a CRL4B ligase complex. Notably, this interaction is independent of a dedicated substrate receptor, thus functionally segregating this mechanism from all described degraders. Collectively, our data outline a versatile and broadly applicable strategy to identify degraders with nonobvious mechanisms and thus empower future drug discovery efforts.
Conflict of interest statement
C.M.-R. and G.E.W. are listed as inventors of a patent application for glue discovery in neddylation-deficient cellular systems. C.M.-R., S.K. and G.E.W. are listed as inventors of patent applications covering the chemical space of dCeMM2/3/4. M.B., S.K., G.E.W and CeMM are founders and equity holders of Proxygen.
Figures












Comment in
-
Prospecting for molecular glues.Nat Chem Biol. 2020 Nov;16(11):1157-1158. doi: 10.1038/s41589-020-0620-z. Nat Chem Biol. 2020. PMID: 32747810 No abstract available.
References
-
- Schneekloth JS, Jr, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–54. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
